Все познается в сравнении (сравнительная геномика). Этапы изучения наследственной болезни. Новая культурная ситуация
Исследования пространственной конфигурации ДНК в хромосомах выявили неожиданные, раннее не известные причины возникновения тяжелых заболеваний человека.
Возникновение трехмерной геномики
На протяжении десятков лет, прошедших с момента доказательства генетической функции ДНК в сороковых годах прошлого века, неизменными оставались представления о том, что мерой расстояния между любыми участками генома является протяженность разделяющей их цепочки ДНК. Сегодня мы знаем, что способность ДНК образовывать петли и другие сложные структуры дает возможность генам и элементам генома, управляющим их работой (энхансерам), оказываться поблизости друг от друга в пространстве клеточного ядра даже в том случае, если они разделены протяженным фрагментом ДНК (рис. 1).
В последние годы появились новые подходы, позволяющие изучать укладку геномной ДНК в клеточном ядре. Это положило начало развитию научного направления, которое мы называем 3D-геномикой. С использованием этих подходов было показано, что хромосомы разделены на структурно-функциональные блоки - топологически-ассоциированные домены (ТАДы). Участки генома из одного ТАДа контактируют друг с другом гораздо чаще, чем с участками из соседних ТАДов. Это позволяет представить ТАДы в виде относительно плотных клубков нити ДНК. Результаты многих экспериментов показывают, что энхансер может активировать только гены, расположенные внутри того ТАДа, где находится его энхансер.

Таким образом, ТАДы играют важную роль в управлении активностью генов. Удаление или повреждение участка ДНК, разделяющего соседние ТАДы, приводит к тому, что энхансер получает возможность активировать гены, которые в норме в данном типе клеток не работают, что может стать причиной возникновения тяжелых заболеваний, таких как рак, нарушения формирования половых признаков и сбои в развитии эмбриона (рис. 2).

Где проходят границы между ТАДами
Но что обеспечивает разделение генома на ТАДы? В решение этой проблемы существенный вклад внесла работа нашей лаборатории. Мы обнаружили, что организация геномной ДНК в ТАДы происходит в значительной мере самопроизвольно и регулируется простыми физическими законами. Наша работа была опубликована в престижном международном журнале Genome Research (Sergey V. Ulianov et al. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains // Genome Research , 2016, 26, p. 70–84, doi: 10.1101/gr.196006.115 ), о ней много говорилось в прессе и по телевидению.
Суть полученных нами результатов заключается в том, что границами ТАДов являются участки генома, содержащие гены «домашнего хозяйства», то есть гены, которые работают во всех типах клеток и необходимы для поддержания базовых клеточных процессов. В силу ряда особенностей такие участки генома не могут сворачиваться в плотные глобулы, тем самым создавая «разметку» границ ТАДов в геноме.
Важно отметить, что помимо разнообразных биохимических техник мы использовали моделирование структуры генома на суперкомпьютере МГУ «Ломоносов», и результаты этого моделирования ясно свидетельствуют о том, что укладка ДНК в индивидуальных клетках может достаточно сильно различаться (рис. 3).
От клеточных популяций к индивидуальным клеткам
В подавляющем большинстве случаев в молекулярно-биологических исследованиях приходится использовать сотни тысяч и даже миллионы клеток в каждом эксперименте. Это связано с тем, что в одной клетке очень мало исследуемых молекул, и это крайне затрудняет работу с ними.
Например, количество геномной ДНК в одной клетке человека примерно в сто тысяч миллионов раз меньше одного грамма. Работа с большим количеством клеток приводит к тому, что получаемые в эксперименте результаты, как правило, позволяют установить средние, наиболее типичные значения тех или иных параметров клеточной физиологии. В известном смысле полученную информацию можно уподобить «средней температуре» пациентов по больнице.
Работа с большим количеством клеток, как правило, позволяет установить средние, наиболее типичные значения тех или иных параметров клеточной физиологии, вроде «средней температуры» пациентов по больнице
Безусловно, результаты исследований клеточных популяций позволили установить много важных закономерностей. Однако хорошо известно, что клетки одного типа, выглядящие под микроскопом совершенно одинаково, могут различаться по множеству разных биохимических параметров. Исследования работы генома в одной отдельно взятой клетке становятся «трендом времени» и уже внесли значительный вклад в понимание того, как осуществляется тонкая настройка работы нашего генома. Такие исследования влияют и на развитие медицины, поскольку, например, события, происходящие в очень малой доле клеток, могут давать старт развитию опухолей. При изучении больших клеточных популяций такие события часто остаются незамеченными.
В совместной работе с австрийскими и американскими коллегами мы разработали новый экспериментальный подход, позволяющий анализировать укладку генома в индивидуальных клетках. С использованием этого подхода нам удалось построить существенно более детализированные карты пространственной организации генома мыши, чем в предшествующей работе английских коллег. Анализ полученных данных, недавно опубликованный в журнале Nature (Ilya M. Flyamer et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition // Nature , 544, p. 110–114, doi: 10.1038/nature21711 ), предоставил веские доказательства того, что укладка генома существенно различается в индивидуальных клетках (рис. 4). По нашему мнению, это свидетельствует о том, что в клетке происходит постоянный перебор различных геномных конфигураций - а это обеспечивает возможность быстрой адаптации к изменяющимся условиям внешней среды.

Хотя в большинстве случаев проще изучать клеточную популяцию, чем индивидуальные клетки, для некоторых типов клеток популяционный подход вообще невозможно использовать, потому что эти клетки являются, что называется, штучным товаром. С использованием разработанного нами экспериментального подхода нам удалось изучить укладку отцовского и материнского геномов в оплодотворенных яйцеклетках (зиготах) мыши.
Совершенно неожиданно для себя мы обнаружили, что укладка геномной ДНК в материнском ядре в зиготе принципиально отличается от укладки генома в ядрах любого другого типа клеток. В ядрах всех прочих исследованных клеточных типов активные и «молчащие» области генома пространственно обособлены друг от друга. В материнском ядре зиготы же, напротив, этого не наблюдается. Наши результаты позволяют предположить, что конфигурация генома в материнском ядре является наиболее базовой, соответствующей так называемому состоянию тотипотентности, позволяющему в ходе эмбрионального развития из одной зиготы получить множество разных клеточных типов взрослого организма.
Пространственная конфигурация генома в материнском ядре является наиболее базовой и позволяет из одной оплодотворенной яйцеклетки получить множество разных типов клеток взрослого организма
3D-геномика и медицина
Обсуждая новости молекулярной биологии, как правило, говорят о «геноме человека», или «геномной ДНК человека», или просто о ДНК. Но важно помнить, что в ядрах клеток нашего организма в норме содержится 23 разных молекулы ДНК, каждая из которых формирует отдельную хромосому, а все вместе они называются геномом.
Каждая хромосома уложена определенным, уникальным для нее способом и располагается в ядре клетки так, что территория, ею занимаемая, практически не пересекается с территориями соседних хромосом. В этом смысле ядро клетки напоминает земной шар, на котором есть много государств, занимающих определенные территории и разделенные границами.
История знает множество примеров того, как события в одном государстве напрямую влияли на жизнь в соседних странах и на мировую политику в целом. В ядре клетки ситуация примерно такая же. Любые изменения в работе генома, будь то запуск или подавление экспрессии отдельных генов, или появление лишних копий тех или иных хромосом, могут повлиять на работу генов, напрямую не затронутых этими изменениями и расположенных в других хромосомах-государствах.
В качестве примера можно указать на результаты работы, которую мы выполнили с нашими французскими коллегами из Института Гюстава Русси. Результаты этой работы были опубликованы в престижном гематологическом журнале Blood (Jeanne Allinne et al. Perinucleolar relocalization and nucleolin as crucial events in the transcriptional activation of key genes in mantle cell lymphoma // Blood , V. 123, 13, p. 2044–2053, doi: 10.1182/blood-2013-06-510511 ). Мы убедительно продемонстрировали, что простое перемещение определенного гена из одной области ядра в другую может быть причиной его активации в клетках, где в норме он не работает. Это запускает целый каскад процессов, который в конечном итоге ведет к развитию лейкоза, первопричины которого было бы сложно понять без учета пространственной структуры генома.
Простое перемещение определенного гена из одной области ядра в другую может запустить целый каскад процессов, который в конечном итоге ведет к развитию лейкоза
Важно отметить, что раскрытие принципиально нового механизма возникновения лейкозов создает базис для разработки путей борьбы с этими заболеваниями. Таким образом, исследования укладки геномной ДНК в ядре представляют интерес не только для фундаментальной науки, но и для медицины, способствуя более глубокому пониманию механизмов возникновения различных патологий.
Эволюция 3D-организации генома
Поскольку трехмерная организация генома является одним из инструментов регуляции экспрессии генов, она должна быть объектом эволюции. В недавно выполненной в нашей лаборатории работе, результаты которой опубликованы в высокорейтинговом международном журнале (Anastasia P. Kovina et al. Evolution of the Genome 3D Organization: Comparison of Fused and Segregated Globin Gene Clusters // Molecular Biology and Evolution , V. 34, 6, p. 1492–1504, doi: 10.1093/molbev/msx100 ), мы показали, что это действительно так.
На примере эволюции кластеров глобиновых генов позвоночных животных мы продемонстрировали, что по мере их продвижения по эволюционной лестнице происходит утрата линейных сегментов хромосом, тогда как сегменты, организованные в глобулы (клубки), сохраняются (рис. 5).
Скорее всего, это связано с тем, что у млекопитающих существенно возрастает роль удаленных энхансеров в регуляции активности генов. Установление контактов между такими энхансерами и подконтрольными им генами обеспечивается за счет образования петель ДНК, что и приводит к образованию глобул.
У позвоночных животных по мере их продвижения по эволюционной лестнице происходит утрата линейных сегментов хромосом, тогда как сегменты, организованные в глобулы (клубки), сохраняются
Заключительные заметки
В последние годы отечественную науку часто и во многих случаях обоснованно критикуют за низкую продуктивность и отсутствие работ международного уровня. Выше мы показали, как одна сравнительно небольшая отечественная лаборатория успешно работает на переднем крае мировой науки, систематически публикуя результаты своей работы в наиболее престижных международных журналах.
Выполнение всех перечисленных выше работ стало возможным благодаря большому гранту Российского научного фонда. Значение такой поддержки трудно переоценить не только потому, что она обеспечивает возможность выполнения дорогостоящих работ, таких как массированное секвенирование ДНК. Но еще важнее то, что такие гранты обеспечивают возможность привлечения к работе молодых исследователей, предоставляя разумную альтернативу отъезду за рубеж. По крайней мере в экспериментальной биологии адресная поддержка коллективов, работающих на мировом уровне (о чем можно судить по наличию публикаций в рейтинговых международных журналах), является, на наш взгляд, наиболее прямым путем к возрождению науки в нашей стране.
(на английском языке - genomics) - это наука, занимающаяся изучением геномов. Количество геномной информации резко возросло в последние годы в связи с улучшением технологии секвенирования ДНК. GenBank, база данных NIH (Национальные Институты Здоровья в США), на апрель 2011 года содержит 135 440 924 секвенированных ДНК.
1956 год стал основополагающим в процессе исследований генетики человека, поскольку в этом году была создана наука хромосология, и проведен съезд по генетике человека в Копенгагене.
Эволюция любой науки обусловлена уточнением моделей и теорий, но при этом новые предположения не отменяют старые истины, так что то, что было верно вчера, не обязательно ложно сегодня. Только псевдо науки являются неизменными веками и гордятся этим, как будто это является своего рода гарантией качества.
Нас осаждают со всех сторон многочисленные дисциплины, старые и новые, которые преподают медицинские практики с исключительными результатами, революционные устройства измерения отрицательных и положительных способностей.
В настоящее время не существует сектора в науке, который не исследовался где то и кем-то в мире: каждый день, научно-исследовательские центры-гиганты при университетах, частные институты и даже мелкие лаборатории, распространяют огромное количество новой информации о новейших исследованиях и дополнений к ним. Иногда эта информация довольно эксцентрична, например в таких секторах как невидимость, сексуальное поведение мух в Китае или молекулярный вес запахов, и в областях, которые оставляют место для увлекательных сценариев, например относящиеся к конструированию жизни в лаборатории или открытия новых планет, которые могли бы принять эту новую жизнь.
Пионером гонки за увеличением срока жизни человека является Крейг Вентер, генетик, предприниматель и филантроп, который стоит за проектом "Геном Человека", в марте этого года заявил, что его последний проект в области геномики, будет использовать капитал в 70 млн. долларов, чтобы создать новую компанию с названием Human Longevity Inc (HLI). Вентер не одинок в своих амбициях. Например, компания Calicо (Калифорнийская компания жизни) имеет цели, улучшения здоровья людей, решения проблемы старения и ассоциативных болезней, и Университет Калифорнии, Сан-Диего - где будут секционировать раковый геном и опухоли HLI всем пациентам, страдающим от рака и кто даст на это свое согласие.
С момента первого секвенирования в 2011 году, геномика быстро прогрессировала, и в настоящее время ученые, изучающие рак будут переходить на новый уровень "следующий рубеж в науке", говорит Липман, директор института Калифорнии. "Сейчас мы находимся в периоде, который будет приравнен исторически для геномики секционирования клеток рака, к периоду 90-х годов для развития интернета. Мы изучаем геном и технологии секционирования в надежде, что по этой шкале могут быть достигнуты быстрые результаты. На что раньше уходило 15-20 лет теперь реально можно будет достигнуть за 1-2 года. Борьба против рака быстро развивается, и это только верхушка айсберга ".
Факты из области геномики:
. В апреле 2003 года, «Проект генома человека» - был завершен после 13 лет исследований. В этот проект было инвестировано 2,7 млрд. долларов.
. В декабре 2005 года был запущен пилотный проект «Атлас генома рака», сроком на 3 года и стоимостью 100 миллионов долларов, предназначенный для изучения генетических соединений клеток рака.
. В мае 2007 года, геном Джеймса Уотсона, один из первооткрывателей ДНК, был "секвенирован" полностью, стоимость операции составила до одного миллиона долларов.
. С конца прошлого года, компания 23andMe предоставляет возможность определения последовательности генома (секвенирование) по цене всего 1000 $.
. В настоящее время, проект генома человека продолжается. После секвенирования обнаружено было около трех миллиардов пар оснований, которые составляют ДНК. Проект ENCODE (Энциклопедия элементов ДНК), родился как результат международного сотрудничества более чем 80 стран и 35 научно-исследовательских групп, он обещает первую интерпретацию информации для описания поведения генома.
Исследователи смогли понять, как и где определенные биологические функции возникают, бросая вызов различным догмам и переоценкам того, что до вчерашнего дня считалось "нежелательной" ДНК или не кодированным (неактивным) ДНК. "Новые данные показывают, что геном содержит очень мало разделов которые не используется",- утверждается в заявлении «Консорциума и Европейской лаборатории молекулярной биологии» (EMBL-EBI), который возглавлял исследование вместе с «Национальным научно-исследовательским институтом генома человека» (NHGRI), «Национальный институт здоровья» (NIH) в США. Опровержение мифа о генетической детерминированности, совершенное проектом « Геном человека», знаменует начало новой постгеномной эры.
Новая культурная ситуация
 До недавнего времени "дизайн" человека, то есть создание всех его характеристик было возложено на природу, никто не мог вмешаться, чтобы улучшить человеческое существо.
До недавнего времени "дизайн" человека, то есть создание всех его характеристик было возложено на природу, никто не мог вмешаться, чтобы улучшить человеческое существо.
Каждый новый организм рождается из маленькой ячейки. Он наследует программу предков в форме ДНК, но не наследует физические тела своих предков. Он наследует сердце родителей, но у него новое сердце. Все начинается с нуля, с одной клетки, но с каждой новой жизнью программа ДНК может получить как улучшения, так и ухудшения.
Прежде чем оценить эффект геномики следует отметить, что было бы невозможно, и даже безответственно, отказаться от генетических методов манипуляции только потому, что эти методы могут быть использованы недобросовестными и эгоистическими людьми в своих целях.
Ни один государственный орган не имеет ту волшебную палочку, которая может заставить исчезнуть все технологии геномики. Главный вопрос развития геномики состоит не в том, чтобы думать, как блокировать этот прогресс, но скорее, как получить максимальную выгоду и минимизировать риски.
Оценка возможностей геномики в целях терапевтических возможностей и в области улучшения генетического фона зависят от этических принципов, которые будут приниматься в качестве ориентира.
Для тех, кто является сторонниками репродукции человека "под наблюдением", и которые готовы принять как факт, возможности использования искусственных методов будет очень легко принять и генетические манипуляции, но для кого-то, это будет неприемлемо.
Выходя за рамки принципов, от которых отталкивается наука, человечество должно иметь в виду, что все технологии геномики применяемые на человеке, имеют на первом плане самого человека. Этот фактор поднимает много знаков вопроса, в том числе и вопрос о том какой эффект может иметь генная инженерия в балансе экосистемы и морали самого человека, который в итоге и является бенефициаром такой науки как геномика.
Прежде чем говорить непосредственно о последствиях, которые могут иметь генетические манипуляции, уточняем, что желание улучшить дизайн человеческого существа, до рождения, это в первую очередь прямое влияние на селекцию, то есть "удаление того что отличается, что не совершенно, неудачно получилось ". Это тоже самое что выбросить неудачный эмбрион в мусор во время процедуры ЭКО.
В среде такой науки как геномика, мы можем говорить о возможности основать новый вид услуг, «ген служба», которая должна будет удовлетворить человеческое желание улучшить свой генофонд. Это услуга, скорее всего, будет платной с поддержкой государства или же строго коммерческой, где каждый человек, при условии, что он платежеспособен, сможет корректировать свою генную информацию.
Но существование этого "сервиса" будет невозможным без технического прогресса и некоторого изменения менталитета человека.
Как и любое лекарство, так и новые технологии геномики могут быть использованы для "сыворотки к гену", где существуют короткосрочные или долгосрочные риски. Всегда присутствует риск, что будут устранены гены, которые имеют еще не известные положительные аспекты и которые могут проявляться в различной среде. Например, тот же ген, который вызывает серповидно-клеточную анемию, делает организм более устойчивым к малярии.
Что касается генной терапии, мы должны предполагать изменения в половых клетках, как следствие соматической генной терапии. В определенных обстоятельствах это может быть законным (следует оценивать, можно ли позволять таким людям, размножаться, после лечения или нет), поскольку модификация половых клеток для лечения может привести к изменениям генетического наследия будущих поколений. Так же развивается генная терапия зародышей и появляется необходимость проводить эксперименты на эмбрионах. Естественно, что до достижения успеха в этих исследования, будет множество неудач, что предполагает то, что объект исследования умрет. Да, во имя науки и во блага будущих поколений можно эти жертвы оправдать, но это не может быть оправдано с этической точки зрения.
Между двумя представителями рода человеческого сходства меньше, чем между двумя различными животными.
Мишель де Монтень
То, что ново в себе, будет понято только по аналогии со старым.
Как уже говорилось, сравнительный метод служит традиционным подходом в старых классических областях биологии (анатомия, эмбриология, цитология). Так, еще Дарвин свою точку зрения о происхождении человека обосновывал с помощью сравнительно-эволюционного метода, указывающего на многочисленное сходство в анатомии и физиологии человека и обезьян.
В последнее время сравнительный подход стал широко и весьма эффективно использоваться в молекулярной биологии и генетике. Мощный толчок этому был дан крупномасштабным секвенированием геномов. Появилось даже новое направление в геномике — сравнительная геномика — сопоставление отдельных генов, групп генов и целых локусов далеко эволюционно отстоящих организмов. Это принципиально важное направление исследований позволяет по-новому решать ряд ключевых вопросов. Рассмотрим некоторые из них.
В настоящее время человечество кроме своей собственной Энциклопедии располагает подобными Энциклопедиями некоторых простейших организмов: кишечной палочки, мухи дрозофилы, дрожжей и червя Caenoharbditis elegans , а также мыши — и отдельными главами из Энциклопедий некоторых других высокоорганизованных организмов (обезьяны, крысы). Сегодня параллельно с секвенированием генома человека идет расшифровка еще около 1000 геномов других животных и растений. ДНКовый текст во всех этих Энциклопедиях написан одними и теми же четырьмя буквами, число которых у бактерий составляет миллионы, у птиц — сотни миллионов и миллиарды у млекопитающих и человека. Поскольку все тексты написаны одинаково, их удается сравнивать между собой. При этом выяснилось, что, несмотря на огромные различия в размерах геномов, число генов (наиболее значимых предложений в текстах) у разных видов организмов не сильно отличается. В этой связи стали говорить даже о неком парадоксе, который получил специальное название G-парадокса (первая буква англ. слова gene — ген). Сейчас этот парадокс объясняют тем, что главное для организма все-таки не общее число генов, а то, как они устроены и как регулируются, какова сложность взаимодействия между продуктами разных генов. «У нас одинаковые гены с кошками и собаками, но они по-разному регулируются», — заявил по этому поводу Крег Вентер, один из главных героев секвенирования человеческого генома. Скорее всего, именно устройство и регуляция работы генов уникальны для человека, делая его «венцом природы». Короче говоря, если ген — это короткое предложение, то из сочетания одних и тех же слов и предложений можно написать как умнейший трактат, так и примитивные детские стишки. Кроме того, важно, как они будут читаться и звучать.
Какими бы уникальными мы не казались сами себе, в нашей ДНК есть довольно много сходства не только с обезьянами и мышами, но даже с маленьким червем C. elegans и мухой дрозофилой. Можно удивляться, но у нас около 50 % генов сходны с таковыми у червя. У человека и мыши еще больше одинаковых генов, хотя в эволюции человек и мышь разошлись уже около 100 миллионов лет назад. В геноме человека на сегодняшний день обнаружено лишь около 300 генов, которых нет у мыши, а общее их число примерно одинаковое. Таким образом, около 99 % генов человека соответствуют генам мыши, причем примерно 80 % из них почти полностью идентичны. Кроме того, до 90 % генов, ответственных за возникновение различных заболеваний, у человека и мыши сходны. Есть, разумеется, и небольшие различия. Так, у мыши гораздо больше генов, отвечающих за обоняние.
Что же касается наших ближайших родственников, то здесь различия еще меньше. Согласно последним данным, в целом геном человека отличается от генома шимпанзе всего лишь максимум на 5 %! Удивительно, но некоторые группы генов (например, гены, ответственные за формирование тела организма) у человека сродни аналогичным группам у биологических видов, возникших еще пятьсот — шестьсот миллионов лет тому назад, во времена так называемого Кембрийского биологического взрыва. Сейчас с нетерпением ожидается тот момент, когда будет полностью секвенирован геном шимпанзе. После этого в сравнительно геномике должен начаться новый очень важный этап. В результате такого сравнения могут быть обнаружены функционально важные мутации, специфические для человека как вида, что в свою очередь откроет новые пути для медицины. Безусловно, эти данные будут также способствовать более полному пониманию процесса эволюции человека.
Сравнения последовательностей ДНК человека с ДНК других организмов уже оказалось очень плодотворным методом поиска новых функционально важных последовательностей в геноме человека. Такой подход был использован и продолжает использоваться для выявления у человека новых белок-кодирующих и не кодирующих белок генов, а также для идентификации потенциальных регуляторных элементов и выяснения механизмов функционировании разных генных наборов. Для этой цели сейчас уже созданы специальные компьютерные программы, позволяющие «вылавливать» в разных геномах эволюционно консервативные области. Все это принципиально важно, поскольку, как уже подчеркивалось выше, мы не можем ставить генетические эксперименты на человеке, но, благодаря сравнительному методу, имеем возможность интерполировать на человека результаты, которые получаются при молекулярно-генетических исследованиях, проводимых на животных.
Так, в силу подобия геномов даже муха дрозофила может быть использована для более полного понимания функций тех или иных человеческих генов, в частности, ответственных за некоторые заболеваний человека. Примером тому может служить изучение гена dFMR-1 мухи, который имеет гомологию с соответствующим геном человека, определяющим синдром ломкости X-хромосомы — тяжелое наследственное нейродегенеративное заболевание. Это исследование позволило заключить, что причина синдрома скорее всего связана с нарушением механизма РНК-интерференции, о котором мы уже говорили выше. И это серьезная «подсказка» для ученых, решающих проблему синдрома ломкости X-хромосомы у людей.
Важно отметить, что когда мы изучаем геном человека, то фактически мы познаем весь живой мир. Геном человека устроен необычайно сложно. Геномы животных и растений чаще всего значительно проще. Поэтому, когда мы узнаем устройство сложного генома, нам будет очень легко от него перейти к изучению простого. А это сулит революцию в таких областях, как ветеринария, селекция растений и животных.
Сравнительная геномика дала ученым новый подход к пониманию вроде бы навсегда скрытого во мраке веков процесса эволюции и его механизмов. Так, например, проведенные сравнения геномов разных видов животных и человека показали наличие определенных тенденций в эволюции. Одна из них заключается в увеличении количества интронов в процессе эволюционного развития у человека, то есть эволюция как бы сопряжена с «разбиением» генома на отдельные функционально значимые фрагменты: на единицу длины ДНК приходится все меньше информации о структуре белков и РНК (экзоны) и возникает все больше участков, не имеющих пока ясного функционального значения (интроны). Проведенные исследования позволяют считают, что природа совершенствовала млекопитающих не столько посредством умножения разнообразия их генов, сколько путем постепенного копирования, модификации и комбинации уже существующих генов, а также путем изменения регуляции экспрессии генов. Специфика и разнообразие строения и функционирования генетического аппарата велики даже среди эукариот. В то же время существует множество общих принципов и механизмов, и результаты их изучения на одних объектах часто с успехом могут переноситься на другие, включая и человека.
Весьма интересные результаты были получены, в частности, при сравнении распределения по хромосомам сходных последовательностей ДНК человека и других животных. Приведем лишь один пример. Как уже указывалось, между геномами человека и мыши имеется большое сходство. На рис. 37 на цветной вклейке изображено расположение в разных хромосомах мыши сходных сегментов отдельных хромосом человека. Глядя на этот рисунок, мы можем увидеть, что участки одних и тех же хромосом человека распределены во множестве хромосом мыши. Это справедливо и наоборот. А что это значит? Это говорит нам о тех путях, по которым шла эволюция млекопитающих (ведь мышь и человек млекопитающие). Тщательно проанализировав картину, изображенную на рис. 37, ученые установили, что на границах разных участков ДНК мыши, которые обнаруживаются в составе ДНК человека, содержатся различные подвижные генетические элементы, тандемные повторы и другие «горячие точки», по которым, вероятно, и шла перестройка (рекомбинация) в ходе многовекового процесса эволюции животных организмов.
Рис. 37 . Генетическое сходство (гомология) хромосом человека и мыши. Разными цветами и номерами на хромосомах мыши отмечены нуклеотидные последовательности человеческих хромосом, содержащие сходные сегмент.
Сравнительная геномика показала, что гены, одинаковые по эволюционному происхождению и выполняемой функции (гомологичные), часто оказываются сцепленными с одними и теми же гомологичными генами у разных видов. На основании этого предсказывают вероятный район локализации генов у одних видов, если известно, с какими генами они сцеплены у других, т. е. проводят «сравнительное картирование». Все это важно в связи с тем, что правила чисел и относительное положение генов на хромосоме не всегда предопределяют законы их функционирования. Так, белковый состав многих специализированных клеток мыши, крысы и человека выглядит похожим, хотя сами гены разбросаны на хромосомах по-разному.
Итак, сравнительная геномика позволяет нам судить о механизмах и путях эволюции геномов и даже на новом уровне воссоздавать классификацию всего животного мира. Все это и есть предмет еще одного нового направления — эволюционной геномики. Ее венцом должно стать создание определенной четкой системы живых организмов, в некотором смысле подобной таблице Менделеева.
Благодаря использованию методов и подходов сравнительной и эволюционной геномики уже получены сенсационные результаты, касающиеся такого сложного и интересного вопроса, как происхождение человека и эволюция его генома. Подробнее об этом и пойдет речь в следующей части книги.
| |
Немного фактов на грани с фантастикой
Часть III. Происхождение и эволюция генома человека
Кафедра медицинской биологии и генетики
Геном. Геномика.
Казань, 2005
Рассматриваемые вопросы
1. Введение в геномику.
1.2. Разделы геномики
1.3. Этапы развития геномики.
2. Организация генома человека. Явление полиморфизма.
2.1. Уникальные гены.
2.2. Семейство генов.
2.3. Регуляторные зоны.
2.4. Повторы.
2.5. Транспозоны.
2.5.1. Биологическое значение транспозируемых элементов.
3. Явление полиморфизма.
1. Введение в геномику.
1.1. Определение генома и геномики.
Прежде всего, определим понятие «геном». Существует несколько определений генома. В энциклопедическом словаре «Генетика» Н.А.Картель и др. даётся два определения генома. Во-первых, под геномом понимают совокупность гаплоидного набора хромосом данного вида организмов. И, во-вторых, - это весь генетический материал отдельного вируса, клетки или организма не являющегося аллоплоидным. В нашем изложении мы будем исходить из того, что геном клетки это вся совокупность ДНК, находящаяся в ядре и митохондриях (пластидах) этой клетки или организма. Такое определение часто используется в работах связанных с изучением генома.
Строение и функцию генома изучает специальная наука – геномика .
Успехи в изучении генома человека стали наиболее ощутимы в связи с разработкой и последующем выполнением международного проекта «Геном человека». Этот международный проект объединил усилия сотен учёных из разных стран и осуществлялся с 1989 г по 2005 г. Главные направления проекта – картирование генов (определение локализации генов в хромосомах) и секвенирование ДНК или РНК (порядок расположения в ДНК или РНК нуклеотидов). Инициатором этого движения с самого начала стал лауреат Нобелевской премии учёный Дж. Уотсон. В России таким энтузиастом стал академик Баев А.А. На проект было затрачено свыше 6 млрд долларов. Материальные затраты России были настолько скромными, что их не учитывают при общем подсчёте издержек. Несмотря на это российские учёные проводили исследования по картированию 3,4,13 и 19 хромосоме. Проект позволил полностью расшифровать последовательность нуклеотидов в геноме человека. Фактически это был первый этап – структурный. Второй этап, который назвали функциональный, будет связан с расшифровкой функции гена. Полученные результаты в области исследования генома легли в основы выпущенного в США Ч. Кэнтором и К. Смит в 2000 году первого учебника для ВУЗов «Геномика».
1.2. Разделы геномики
Геномика подразделяется на пять самостоятельных разделов.
Структурная геномика изучает последовательность нуклеотидов в геноме, определяет границы и строение генов, межгенных участков, промоторов, энхансеров и др., т.е. фактически принимает участие в составлениигенетических карты организма. Подсчитано, что геном человека состоит из3,2 млрд нуклеотидов.
Функциональная геномика идентифицирует функцию каждого гена и участка генома, их взаимодействие в клеточной системе. Одна из важнейших задач геномики создать, так называемую«генную сеть» - взаимосвязанную работу генов. Например, генная сеть системы кроветворения включает в себя работу не менее 500 генов. Они не только взаимосвязаны между собой, но связаны и с другими генами.
Сравнительная геномика изучает сходства и различия в организации геномов разных организмов.
Эволюционная геномика объясняет пути эволюции геномов, происхождение генетического полиморфизма и биоразнообразия, роль горизонтального переноса генов. В применении к человеку, также как и к любому организму, можно сказать, что эволюция человека – это эволюция генома.
Медицинская геномика решает прикладные вопросы клинической и профилактической медицины на основе знания геномов человека и патогенных организмов.
Геномика человека является основой молекулярной медицины и её достижения используются при разработке эффективных методов диагностики, лечения и профилактики наследственных и не наследственных заболеваний. Если раньше предполагали, что наследственная патология, связана с определёнными генами или регуляторными зонами, то сейчас, всё большее внимание привлекают нуклеотидные последовательности, располагающиеся в межгенных промежутках. Они долгое время считались «молчащими». В настоящее время накапливается всё больше сведений об их влиянии на экспрессию генов.
Исследования в области генома ещё раз подтвердили необходимость индивидуального подхода к профилактике и лечению заболеваний. Значительный интерес представляют для медицины исследования связанные с составлением «генной сети» - схем взаимодействия генов между собой на уровне белковых продуктов. Эти исследования способствовали созданию в рамках геномики новой науки – протеомики , которая изучает белковый пейзаж клетки в различных режимах функционирования генов. Полученные результаты однозначно показывают целесообразность индивидуального подхода к лечению заболевания. Сейчас протеомика – самостоятельная наука, тесно связанная с геномикой.
В этой связи следует подчеркнуть, что тезис «лечить не болезнь, а больного» получил существенное подтверждение в многочисленных исследованиях генома и белков. Основываясь на них приоритетность этого положения в медицинской практике перестала вызывать сомнения.
Это часть 1 истории геномики, которая называется "геномные проекты". В этой части я постараюсь научно-популярно рассказывать о том, как появились первые методы чтения генетических последовательностей, в чем они заключались и как геномика двигалась от чтения отдельных генов к чтению полных геномов, в том числе полных геномов конкретных людей.
Вскоре после открытия Уотсона и Крика (Рис.1) рождается наука геномика. Геномика - это наука об исследовании геномов организмов, которая включает интенсивное чтение полных последовательностей ДНК (секвенирование) и их нанесение на генетические карты. Это наука так же рассматривает взаимодействия между генами и аллелями генов и их разнообразие, закономерности в эволюции и устройства геномов. Развитие этой области происходило так стремительно, что еще совсем недавно текстовые редакторы вроде Microsoft Word не знали слова “геном” и пытались исправить его на слово “гном”.
Рис. 1
Джеймс Уотсон (слева) и Френсис Крик (справа) - ученые открывшие двойную спираль ДНК
Самый первый прочтенный ген был ген оболочки бактериофага MS2, изученный в лаборатории Валтера Файерса в 1972-ом году . В 1976-ом были известны и другие гены бактериофага - его репликаза, ген отвечающий за размножение вирусных частиц . Короткие молекулы РНК тогда уже читались сравнительно легко, но крупные молекулы ДНК читать толком еще не умели. К примеру, полученная в 1973-ем году Вальтером Гилбертом и Алленом Максам последовательность участка гена лактозного оперона, длинной в 24 буквы, рассматривалась как существенный прорыв в науке. Вот эта последовательность:
5"—TGGAATTGTGAGCGGATAACAATT 3"
3"—ACCTTAACACTCGCCTATTGTTAA 5"
Первые техники чтения ДНК были очень неэффективными и использовали радиоактивные метки для ДНК и химические методы, чтобы различить нуклеотиды. Например, можно было взять ферменты, которые разрезают нуклеотидную последовательность с разной вероятностью после разных букв. Молекула ДНК состоит из 4-ех букв (нуклеотидов) A, T, G и С, которые входят в состав двойной анти-параллельной (две цепи направлены в противоположные стороны) спирали. Внутри этой спирали нуклеотиды находятся друг напротив друга в соответствии с правилом комплементарности: напротив А в другой цепи стоит T, напротив G стоит С и наоборот.
Гилберт и Максам использовали 4 типа ферментов. Один разрезал после А или G, но лучше после A (A>G), второй разрезал лучше после G (G>A), третий после C, а четвертый после С или T (С+T) . Реакция проводилась в 4-ех пробирках с каждым типом ферментов, а затем продукты помещали на гель. ДНК - заряженная молекула и при включении тока бежит от минуса к плюсу. Маленькие молекулы бегут быстрее, поэтому разрезанные молекулы ДНК выстраиваются по длине. Глядя на 4 дорожки геля, можно было сказать в какой последовательности расположены нуклеотиды.
Прорыв в области секвенирования ДНК случился, когда английский биохимик Фредерик Сенгер в 1975-ом году предложил, так называемый “метод терминации цепи” для чтения последовательностей ДНК. Но прежде чем рассказать об этом методе, необходимо ввести в курс процессов происходящих при синтезе новых молекул ДНК. Для синтеза ДНК необходим фермент - ДНК-зависимая ДНК полимераза, которая способен достраивать одноцепочечную молекулу ДНК до двухцепочечной. Для этого ферменту необходима “затравка” - праймер, короткая последовательность ДНК, способная связаться с длинной одноцепочечной молекулой, которую мы хотим достроить до двухцепочечной. Так же необходимы сами нуклеотиды в форме нуклеотидтрифосфатов и некоторые условия, такие как определенное содержание ионов магния в среде и определенная температура. Синтез всегда идет в одном направлении от конца называемого 5’ к концу называемому 3’. Разумеется, для чтения ДНК необходимо большое количество матрицы - то есть копий той ДНК, которую собираются читать.
В 1975-ом году Сенгер придумал следующее. Он брал специальные (терминирующие) нуклеотиды, которые, присоединившись к растущей цепи молекулы ДНК, мешали присоединению последующих нуклеотидов, то есть “обрывали” цепь. Далее он брал 4 пробирки, в каждую из которых добавлял все 4 типа нуклеотидов и один тип терминирующих нуклеотидов в небольшом количестве . Таким образом, в пробирке, где находился терминирующий нуклеотид “А” синтез каждой новой молекулы ДНК мог оборваться в любом месте, где должна была встать “А”, в пробирке с терминирующей “G” - в любом месте, где должна встать G и так далее. На гель наносились 4 дорожки из 4-ех пробирок (Рис. 2) и снова самые коротки молекулы “убегали” вперед, а самые длинные оставались в начале, а по отличиям в полосах можно было сказать, какой нуклеотид следует за каким. Чтобы увидеть полосы, один из четырех нуклеотидов (A, T, G или C) метился, без изменения химических свойств, с использованием радиоактивных изотопов.
Рис. 2 Метод Сангера. Показаны три серии из 4-ех дорожек.
С помощью этого метода был прочитан первый геном, основанный на ДНК - геном бактериофага ϕX174, длинной 5,386 нуклеотидов (геном фага MS2, прочитанный ранее был на основе РНК и имел геном длинной 3,569 нуклеотидов).
Метод Сенгера был существенно улучшен в лаборатории Лероя Худа, где в 1985-ом году радиоактивную метку смогли заменить светящейся, флюрисцентной меткой . Это дало возможность создать первый автоматический секвенатор: каждая молекула ДНК теперь была покрашена разным цветом в зависимости от того, какой была последняя буква (меченый цветом нуклеотид, обрывающий цепь). Фрагменты разделялись на геле по размерам и машина автоматически считывала спектр свечения поступающих полос, выдавая результаты на компьютер. В результате такой процедуры получается хроматограмма (Рис. 2), по которой легко установить последовательность ДНК длинной до 1000 букв, с очень небольшим количеством ошибок.
Рис. 3 Пример хроматограммы, на современном секвенторе, использующий метод обрывания цепи Сангера и светящуюся метку.
На многие годы улучшенный метод Сенгера станет основным методом массового секвенирования геномов и будет использован для многих проектов полных геномов, а Сенгер в 1980-ом получит вторую нобелевскую премию по химии (первую он получил еще в 1958-ом за прочтение аминокислотной последовательности белка инсулина - первого прочитанного белка). Первым полным геномом клеточного организма стал геном бактерии, вызывающей некоторые формы пневмонии и менингита - Haemophilus influenzae в 1995-ом году. Геном этой бактерии имел длину 1,830,137 нуклеотидов. В 1998-ом году появляется первый геном многоклеточного животного, круглого червяка Caenorhabditis elegans (Рис. 4 справа), с 98 миллионами нуклеотидов, а затем в 2000-ом году появляется первый растительный геном - Arabidopsis thaliana (Рис. 4 слева), родственницы хрена и горчицы. Геном этого растения имеет длину 157 миллионов нуклеотидов. Скорость и масштабы секвенирования росли с изумительной скоростью и появляющиеся базы данных нуклеотидных последовательностей пополнялись все быстрее и быстрее.
Рис. 4
Arabidopsis thaliana
(слева) и Caenorhabditis elegans
(справа).
Наконец, настал черед генома млекопитающих: геномы мыши и человека. Когда в 1990-ом году Джеймс Уотсон возглавил проект чтения полного генома человека в Институте Национального Здоровья (NIH) в США многие ученые скептически относились к этой идее. Подобный проект требовал колоссальных вложений денег и времени и, учитывая ограниченные возможности существовавших машин для чтения геномов, многим казался просто не выполнимым. С другой стороны проект обещал революционные изменения в медицине и понимании устройства человеческого организма, но и здесь были свои проблемы. Дело в том, что в тот момент не существовало какой-либо точной оценки количества генов у человека. Многие полагали, что сложность устройства человеческого организма указывает на наличие у него сотен тысяч генов, а может и несколько миллионов, а, следовательно, разобраться в таком количестве генов, даже если их последовательности удастся прочитать, будет непосильной задачей. Именно в наличии большого количества генов многие предполагали принципиальное отличие человека от других животных - представление, впоследствии опровергнутое проектом генома человека.
Сама идея прочитать геном человека родилась в 1986-ом году по инициативе Департамента Энергии США, который впоследствии финансировал проект вместе с NIH. Стоимость проекта была оценена в 3 миллиарда долларов, а сам проект был рассчитан на 15 лет при участии в проекте целого ряда стран: Китай, Германия, Франция, Великобритания и Япония. Для чтения генома человека использовались так называемые “искусственные бактериальные хромосомы” (BAC - bacterial artificial chromosome). При этом подходе геном разрезаются на множество частей, длинной примерно в 150000 тысяч нуклеотидов. Эти фрагменты встраивают в искусственные кольцевые хромосомы, которые встраиваются в бактерии. С помощью бактерий эти хромосомы размножаются, и ученые получают множество копий одного и того же фрагмента молекулы ДНК. Каждый такой фрагмент затем читается отдельно, а прочитанные куски по 150000 нуклеотидов наносятся на карту хромосомы. Данный метод позволяет довольно точно секвенировать геном, однако требует очень больших затрат времени.
Но проект генома человека двигался крайне медленными темпами. Ученый Крейг Вентер и его компания Celera Genomics, основанная в 1998-ом году, сыграли примерно такую же роль в истории геномики, как Советский Союз повлиял на полет американцев на луну. Вентер заявил, что его компания закончит проект генома человека раньше, чем завершится государственный проект. На проект потребуется всего 300 миллионов долларов - лишь малую фракцию от затрат государственного проекта, используя новую технологию секвенирования “whole genome shotgun” - чтение случайных коротких фрагментов генома. Когда Френсис Коллинс, сменивший в 1993-ем году Джеймса Уотсона на посту руководителя проекта по чтению генома человека, узнал о намерениях Вентера, он был шокирован. “Мы сделаем геном человека, а вы можете сделать мышку ” - предложил Вентер. Научное сообщество всполошилось, и на то был ряд причин. Во-первых, Вентер обещал закончить свой проект в 2001-ом году, на 4 года раньше срока, намеченного для государственного проекта. Во-вторых, компания Celera Genomics собиралась заработать на проекте, создав абсолютную базу данных, которая была бы платной для коммерческих фармоцевтических компаний.
В 2000-ом году Селера доказала эффективность своего метода секвенирования, опубликовав геном плодовой мушки дрозофилы вместе с лабораторией генетика Джеральда Рубина (ранее whole genome shotgun использовался для прочтения первого генома бактерии, но мало кто верил, что этот метод пригоден для крупных геномов). Именно такой пинок со стороны коммерческой компании стимулировал разработку улучшенных и применение более современных методов чтения геномов в проекте генома человека. В 2001-ом году был опубликован предварительный вариант генома со стороны государственного геномного проекта и Селеры . Тогда была сделана предварительная оценка количества генов в геноме человека, 30-40 тысяч. В 2004-ом году вышла окончательная версия генома , почти на два года раньше, чем следовало по плану. В последней статье было сказано, что количества генов у человека предположительно составляет лишь 20-25 тысяч. Это число сравнимо с другими животными, в частности с червяком C. elegans .
Практически никто не угадал, что количество генов, обеспечивающих работу нашего организма, может быть столь мало. Позже стали известны и другие подробности: геном человека имеет длину около трех миллиардов нуклеотидов, большую часть генома составляют не кодирующие последовательности, в том числе всевозможные повторы. Лишь небольшая часть генома действительно содержит гены - участки ДНК, с которых считываются функциональные молекулы РНК. Интересный факт, что по мере увеличения знаний о геноме человека, число предполагаемых генов только сокращалось: многие потенциальные гены оказывались псевдогенами (не работающими генами), в других случаях несколько генов оказывались частью одного и того же гена.
Дальнейшие темпы секвенирования возрастали экспоненциально. В 2005-ом году опубликован геном Шимпанзе , который подтвердил потрясающее сходство между обезьянами и человеком, которое видели еще зоологи прошлого. К 2008-ому году были полностью прочитаны геномы 32-ух позвоночных, включая кошку, собаку, лошадь, макаку, орангутанга и слона, 3 генома беспозвоночных вторичноротых, 15 геномов насекомых, 7 геномов червяков и сотни геномов бактерий.
Наконец в 2007-ом человечество приблизилась к возможности секвенирования геномов индивидуальных людей. Первым человеком, для которого прочитали полный индивидуальный геном, стал Крейг Вентер (Рис. 4). При этом геном был прочитан так, что можно было сравнить хромосомы Вентера, доставшиеся ему от обоих родителей. Так было выяснено, что между одним и другим набором хромосом внутри одного человека существует около трех миллионов однобуквенных нуклеотидных отличий, не считая огромного количества крупных варьирующих участков. Год спустя опубликован полный диплоидный геном Джеймса Уотсона (Рис. 5). Геном Уотсона содержал 3.3 миллиона однобуквенных замен по сравнению с аннотированным геномом человека, из которых более 10000 вели к изменением в белках, которые кодируют его гены. Геном Уотсона обошелся в 1 миллион долларов, то есть цена на чтение геномов упала более чем в 3000 раз за 10 лет, но и это не предел. Сегодня перед учеными стоит задача ‘1 геном - 1000 $ - 1 день” и она уже не кажется невыполнимой с появлением новых технологий секвенирования. О них расскажет следующая часть "истории".
Рис. 5 Джеймс Уотсон и Крейг Вентер - первые люди с индивидуальными прочитанными геномами.
- Watson J, Crick F: A Structure for Deoxyribose Nucleic Acid . Nature 1953(171):737-738.
- Min Jou W, Haegeman G, Ysebaert M, Fiers W: Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature 1972, 237(5350):82-88.
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A et al: Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 1976, 260(5551):500-507.
- Gilbert W, Maxam A: The nucleotide sequence of the lac operator. Proc Natl Acad Sci U S A 1973, 70(12):3581-3584.
- Maxam AM, Gilbert W: A new method for sequencing DNA. Proc Natl Acad Sci U S A 1977, 74(2):560-564.
- Sanger F, Nicklen S, Coulson AR: DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977, 74(12):5463-5467.
- Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent SB, Hood LE: Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321(6071):674-679.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM et al: Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269(5223):496-512.
- Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 1998, 282(5396):2012-2018.
- Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408(6814):796-815.
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF et al: The genome sequence of Drosophila melanogaster. Science 2000, 287(5461):2185-2195.
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA et al: The sequence of the human genome. Science 2001, 291(5507):1304-1351.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al: Initial sequencing and analysis of the human genome. Nature 2001, 409(6822):860-921.
- Finishing the euchromatic sequence of the human genome. Nature 2004, 431(7011):931-945.
- Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437(7055):69-87.
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G et al: The diploid genome sequence of an individual human. PLoS Biol 2007, 5(10):e254.
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT et al: The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452(7189):872-876.
 Девочка заговорила на иностранном языке после комы
Девочка заговорила на иностранном языке после комы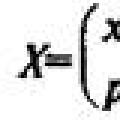 Сравнение законов распределения вероятностей
Сравнение законов распределения вероятностей Советский поэт Асадов Эдуард Аркадьевич: личная жизнь, творчество
Советский поэт Асадов Эдуард Аркадьевич: личная жизнь, творчество