Методы количественного анализа лекарственных средств. Методы исследования лекарственных веществ. В ГФ XI приведены методики установления указанных констант и способы их расчета
Одна из наиболее важных задач фармацевтической химии - это разработка и совершенствование методов оценки качества лекарственных средств.
Для установления чистоты лекарственных веществ используют различные физические, физико-химические, химические методы анализа или их сочетание.
ГФ предлагает следующие методы контроля качества ЛС.
Физические и физико-химические методы. К ним относятся: определение температур плавления и затвердевания, а также температурных пределов перегонки; определение плотности, показателей преломления (рефрактометрия), оптического вращения (поляриметрия); спектрофотометрия - ультрафиолетовая, инфракрасная; фотоколориметрия, эмиссионная и атомно-абсорбционная спектрометрия, флуориметрия, спектроскопия ядерного магнитного резонанса, масс-спектрометрия; хроматография - адсорбционная, распределительная, ионообменная, газовая, высокоэффективная жидкостная; электрофорез (фронтальный, зональный, капиллярный); электрометрические методы (потенциометрическое определение pH, потенциометрическое титрование, амперометрическое титрование, вольтамперометрия).
Кроме того, возможно применение методов, альтернативных фармакопейным, которые иногда имеют более совершенные аналитические характеристики (скорость, точность анализа, автоматизация). В некоторых случаях фармацевтическое предприятие приобретает прибор, в основе использования которого лежит метод, еще не включенный в Фармакопею (например, метод рама- новской спектроскопии - оптический дихроизм). Иногда целесообразно при определении подлинности или испытании на чистоту заменить хроматографическую методику на спектрофотометрическую. Фармакопейный метод определения примесей тяжелых металлов осаждением их в виде сульфидов или тио- ацетамидов обладает рядом недостатков. Для определения примесей тяжелых металлов многие производители внедряют такие физико-химические методы анализа, как атомно-абсорбционная спектрометрия и атомно-эмиссионная спектрометрия с индуктивно связанной плазмой.
Важной физической константой, характеризующей подлинность и степень чистоты ЛС, является температура плавления. Чистое вещество имеет четкую температуру плавления, которая изменяется в присутствии примесей. Для лекарственных веществ, содержащих некоторое количество допустимых примесей, ГФ регламентирует интервал температуры плавления в пределах 2 °С. Но в соответствии с законом Рауля (АТ = iK3C, где АТ - понижение температуры кристаллизации; К3 - криоскопическая постоянная; С - концентрация) при і = 1 (неэлектролит) значение А Г не может быть одинаковым для всех веществ. Это связано не только с содержанием примесей, но и с природой самого ЛВ, т. е. с величиной криоскопической постоянной К3, отражающей молярное понижение температуры плавления ЛВ. Таким образом, при одинаковом АТ = = 2 °С для камфоры (К3 = 40) и фенола (К3 = 7,3) массовые доли примесей не равны и составляют соответственно 0,76 и 2,5 %.
Для веществ, которые плавятся с разложением, обычно указывается температура, при которой вещество разлагается и происходит резкое изменение его вида.
В некоторых частных статьях ГФ X рекомендуется определять температуру затвердевания или температуру кипения (по ГФ XI - «температурные пределы перегонки») для ряда жидких ЛС. Температура кипения должна укладываться в интервал, приведенный в частной статье.
Более широкий интервал свидетельствует о присутствии примесей.
Во многих частных статьях ГФ X приведены допустимые значения плотности, реже вязкости, подтверждающие подлинность и доброкачественность ЛС.
Практически все частные статьи ГФ X нормируют такой показатель качества ЛС, как растворимость в различных растворителях. Присутствие примесей в ЛВ может повлиять на его растворимость, снижая или повышая ее в зависимости от природы примеси.
Критериями чистоты являются также цвет ЛВ и/или прозрачность жидких лекарственных форм.
Определенным критерием чистоты ЛС могут служить такие физические константы, как показатель преломления луча света в растворе испытуемого вещества (рефрактометрия) и удельное вращение, обусловленное способностью ряда веществ или их растворов вращать плоскость поляризации при прохождении через них плоскополяризованного света (поляриметрия). Методы определения этих констант относятся к оптическим методам анализа и применяются также для установления подлинности и количественного анализа ЛС и их лекарственных форм.
Важным критерием доброкачественности целого ряда ЛС является содержание в них воды. Изменение этого показателя (особенно при хранении) может изменить концентрацию действующего вещества, а, следовательно, и фармакологическую активность и сделать ЛС не пригодным к применению.
Химические методы. К ним относятся: качественные реакции на подлинность, растворимость, определение летучих веществ и воды, определение содержания азота в органических соединениях, титриметрические методы (кислотно-основное титрование, титрование в неводных растворителях, комплек- сонометрия), нитритометрия, кислотное число, число омыления, эфирное число, йодное число и др.
Биологические методы. Биологические методы контроля качества ЛС весьма разнообразны. Среди них испытания на токсичность, стерильность, микробиологическую чистоту.
Для проведения физико-химического анализа полупродуктов, субстанций лекарственных средств и готовых лекарственных форм при проверке их качества на соответствие требованиям ФС контрольно-аналитическая лаборатория должна быть оснащена следующим минимальным набором оборудования и приборов:
ИК-спектрофотометр (для определения подлинности);
спектрофотометр для спектрометрии в видимой и УФ-области (определение подлинности, количественное определение, однородность дозирования, растворимость);
оборудование для тонкослойной хроматографии (ТСХ) (определение подлинности, родственных примесей);
хроматограф для высокоэффективной жидкостной хроматографии (ВЭЖХ) (определение подлинности, количественное определение, определение родственных примесей, однородности дозирования, растворимости);
газожидкостной хроматограф (ГЖХ) (содержание примесей, определение однородности дозирования);
поляриметр (определение подлинности, количественное определение);
потенциометр (измерение pH, количественное определение);
атомно-абсорбционный спектрофотометр (элементный анализ тяжелых металлов и неметаллов);
титратор К. Фишера (определение содержания воды);
дериватограф (определение потери массы при высушивании).
Лекция №2по курсу «Анализ и контроль
качества лекарственных средств»
1
Краткий план лекции
1. Классификация ЛВ. Общая характеристикафармакопейного анализа ЛВ. Реактивы, используемые в
фармакопейном анализе.
2. Физико-химические свойства лекарственных веществ
(агрегатное состояние, внешний вид, окраска, кристалличность,
полиморфизм и методы его исследования. Растворимость.
Кислотно-основные свойства лекарственных веществ).
3. Физические константы лекарственных средств и методы
их определения.
4. Методы идентификации лекарственных средств
5. Примеси в лекарственных средствах, классификация,
методы идентификации и анализа. Понятие о стрессовых
испытаниях
6. Методы количественного анализа лекарственных
средств
2
Классификация ЛВ
1. Неорганические вещества (производные s-, p- и dэлементов).2. Органические вещества
2.1. Алифатические соединения (алканы,
галогеналканы, спирты, альдегиды, простые эфиры,
углеводы, аминокислоты, карбоновые кислоты)
2.2. Ароматические соединения (фенолы,
ароматические карбоновые кислоты, ароматические
аминокислоты, фенилалкиламины,
сульфаниламиды);
2.3. Стероидные соединения, простагландины
3
Классификация ЛВ (продолжение)
2.3. Гетероциклические соединения2.3.1. Соединения, содержащие один гетероатом
(производные фурана, бензофурана, пиридина,
хинолина, изохинолина и др.);
2.3.2. Соединения содержащие два и более
одинаковых гетероатома (производные пиразола,
имидазола, бензимидазола, пурина, птеридина и
др.).
2.3.3. Соединения содержащие два и более разных
гетероатомов (производные тиазола, бензотиазола,
оксазолидины и др.).
2.4. Элементорганические вещества.
3. Радиофармацевтические препараты.
4. Биотехнологические (высокомолекулярные)
лекарственные вещества
4
Фармацевтический анализ (анализ ЛВ и ЛС)
Фармацевтический анализ – это раздел науки охимической характеристике и измерении БАВ на всех
этапах производства – от контроля сырья до оценки
качества полученного ЛВ, изучения его стабильности
(установления сроков годности) и стандартизации ЛФ и
ЛС.
Особенности:
1. Проводится анализ совершенно различных по
природе, структуре и свойствам веществ
2. Измеряемые концентрации (содержания) находятся в
диапазоне от 10-9 (1 ppb) до 100%.
3. Анализируются не только индивидуальные ЛВ, но и их
5
смеси.
Фармацевтический анализ (классификации)
В зависимости от поставленных задач:1. Фармакопейный анализ
2. Постадийный контроль производства ЛВ и ЛС
3. Анализ индивидуальных ЛС
4. Аптечный экспресс-анализ
5. Биофармацевтический анализ
В зависимости от результата:
1. Качественный
2. Количественный
3. Полуколичественный (предельные испытания)
6
Критерии фармацевтического анализа
1. Избирательность (специфичность, селективность) –способность однозначно оценивать определяемый
компонент выбранным методом независимо от других
присутствующих веществ (примесей, продуктов распада и
др.) в испытуемом образце в пределах заданного
диапазона применения.
2. Чувствительность
2.1. Предел обнаружения
2.2. Предел определения
3. Правильность – отражение разницы между истинным
содержанием определяемого компонента и
экспериментальным результатом анализа.
4. Воспроизводимость (прецизионность) –
характеристика «рассеивания» результатов возле
среднего значения определяемой величины.
5. Робастность – характеристика устойчивость методики
во времени.
Эти критерии устанавливаются в процессе валидации 7
методов (методик)
Фармакопейный анализ ЛВ (общая структура)
агрегатное состояние,внешний вид,
окраска, кристалличность,
полиморфизм
Подлинность
Первая идентификация
(специфичный метод)
Вторая идентификация
(потверждение)
Определение
физических
констант,
ф/х свойств
Фармакопейный
анализ ЛВ
(общая структура)
температура плавления, температура
затвердевания, температура каплепадения,
температурные пределы перегонки
температура кипения,
плотность и вязкость жидкостей, удельное
вращение и показатель преломления
растворимость, pH
Определение
примесей
Количественное
определение
Показатели микробной чистоты,
стерильность, апирогенность, отсутствие вирусных тел
8
Химическое название
Используется номенклатура IUPAC(International Union Pure Applied Chemistry) – Международный союз
чистой и прикладной химии)
(гораздо реже – тривиальные названия)
1) определяют тип номенклатуры (заместительная, радикальнофункциональная);
2) определяют тип характеристической группы, которую следует принять
за главную;
3) определяют родоначальную структуру (главную цепь, старшую
циклическую систему);
4) дают название исходной структуре и основным группам;
5) дают название префиксам;
6) проводят нумерацию;
7) объединяют частичные названия в общее полное название,
придерживаясь алфавитного порядка для всех определяемых префиксов.
Помимо названия указывают структурную химическую формулу
и брутто-формулу.
9
10. Пример оформления
2-(нафтален-1-илметил)-4,5-дигидро-1Н-имидазолагидрохлорид
10
11. Пример построения химического названия органического ЛВ
Выбор нумерации: от атома азота,ближайшего к старшему заместителю
(С=О-группе).
Установление родоначальной
структуры: 1,4-бензодиазепин;
Название с учетом заместителей: 2,3дигидро-2Н-1,4-бензодиазепин-2-он;
Перечисление заместителей: по
алфавиту – 7-Cl-1-Me-5-Ph
Итого:
7-хлор-1-метил-5-фенил-2,3дигидро-2Н-1,4-бензодиазепин-2-он
H3C
O
N
Cl
N
11
12. Пример построения химического названия органического ЛВ (2)
2-метил-3-гидрокси4,5-ди(гидроксиметил)пиридин
HO
OH
4
3
5
2
HO
6
N
1
12
13. Описание ЛВ
1. Агрегатное состояние (жидкость, газ, твердоевещество, кристалличность), цвет, запах, особые
свойства (гигроскопичность, легкая окисляемость на
воздухе и др.), размер частиц (для тв. веществ).
2. Полиморфизм – явление, характерное для
твердых веществ – способность вещества в твердом
состоянии существовать в различных
кристаллических формах при одном и том же
химическом составе.
При описании сольватов (гидратов) используется
термин «псевдополиморфизм» (изменчивость
состава сольвата или гидрата).
13
14. Описание ЛВ - полиморфизм
Полиморфные формы проявляютодинаковые химические свойства
в растворах и расплавах, но в
твердом состоянии их физические
(плотность, Т плавл, сжимаемость)
и физико-химические свойства
(растворимость и как следствие
биодоступность) могут
существенно различаться.
Та из полиморфных форм,
которая имеет меньшее значение
свободной энтальпии, является
наиболее термодинамически
стабильной, а остальные формы
могут находиться в т.н.
«метастабильном» состоянии. 14
15. Полиморфизм (примеры)
Аллотропные формы углерода: a) лонсдейлит; б) алмаз;в) графит; г) аморфный углерод; д) C60 (фуллерен);
е) графен; ж) однослойная нанотрубка
15
16. Полиморфизм (примеры)
Нимесулид (на формуле показаны торсионные вращения иупаковка, соответствующая полиморфной форме I)
16
17. Полиморфизм (примеры)
Нимесулид (на формуле показаны суммарные торсионныевращения и упаковка, соответствующая полиморфной форме II)
17
18. Полиморфизм (примеры)
Данныерентгеновской
дифракции для
форм I и II
нимесулида
18
19. Полиморфизм (примеры)
Дифференциальная сканирующая калориметрия(DSC) полиморфных форм нимесулида
19
20. Полиморфизм и биодоступность
Кинетика растворения двух полиморфныхформ нимесулида (37С, рН 7,5)
20
21. Методы исследования полиморфных форм
1. Рентгеновская дифракция (порошок икристаллы)
2. Дифференциальная сканирующая
калориметрия, микрокалориметрия
3. Термогравиметрия
4. Анализ поглощения влаги
5. ИК-Фурье-спектроскопия
6. Рамановская спектроскопия
7. Изучение растворимости (кинетики
растворения)
21
22. Размер частиц (порошки, пеллеты)
Для определения размерачастиц использую наборы
сит с квадратными
отверстиями,
изготовленные из инертных
материалов. Степень
измельчения указывается с
использованием номера
сита (размер стороны
отверстия в мкм).
Современные методы – методы
лазерного сканирования
22
23. Растворимость
Данные о растворимости вещества означаютприблизительную растворимость при температуре
20°С, если нет других указаний. Выражение
«растворим в стольких-то частях» следует понимать
как указание на число миллилитров растворителя
(представленное указанным числом частей), в
которых растворим 1 г твердого вещества.
Иногда для обозначения растворимости вещества
используются описательные термины (легко, плохо,
трудно и т.д.).
Классическое описание растворимости (справочники)
– 1 г вещества растворяется в Х г растворителя при
температуре Т.
23
24. Растворимость
2425. Кислотно-основные свойства
Не приводятся в нормативных документах поконтролю качества ЛВ, но имеют решающее
значение при проведении испытаний,
растворимости в водных средах, выборе
методик и методов анализа, а также
всасыванию, распределению,
биодоступности ЛВ.
По кислотно-основным свойствам все
вещества делятся на неионогенные (не
кислота/не основание) и ионогенные –
кислоты (проявляющие в основном
кислотные свойства), основания, амфолиты.
25
26. Методы определения физических констант
1. Гравиметрия2. Рефрактометрия
3. Поляриметрия
4. Вискозиметрия (капиллярная,
ротационная)
5. Термометрия
26
27. Относительная плотность (d20)
Относительная плотность d представляет собой отношениемассы определенного объема вещества к массе равного его
объема воды при температуре 20оС.
Относительную плотность d определяют с помощью
пикнометра, плотномера, гигростатических весов или ареометра
с точностью до десятичных знаков, обозначенных в частной
статье. Атмосферное давление при взвешивании не учитывают,
так как связанная с ним ошибка не превышает единицы в
третьем десятичном знаке.
Кроме того, обычно используют два других определения.
Относительная плотность вещества представляет собой
отношение массы определенного объема вещества при
температуре 20оС к массе равному ему объема воды при
температуре 4оС.
Плотность ρ20 - это отношение массы вещества к его объему
при температуре 20оС. Плотность выражают в килограммах на
кубический метр (1 кг/м3 = 10 –3 г/см3). Чаще всего измерение
плотности выражается в граммах на кубический сантиметр
27
(г/см3).
28. Относительная плотность
2829.
2930. Показатель преломления
3031. Рефрактометры
3132.
3233. Оптическое вращение
3334. Оптическое вращение
3435.
3536. Поляриметрия (оборудование)
3637. Вязкость
Вязкость (внутреннее трение) – свойство текучих тел оказыватьсопротивление передвижению одной их части относительно
другой.
Текучие тела могут иметь ньютоновский тип течения.
Ньютоновскими жидкостями называют системы, вязкость которых
не зависит от напряжения сдвига и является постоянной
величиной в соответствии с законом Ньютона.
Для ньютоновских жидкостей различают динамическую,
кинематическую, относительную, удельную, приведенную и
характеристическую вязкости. Для неньютоновских жидкостей
характерна, главным образом, структурная вязкость.
Динамическая вязкость или коэффициент вязкости η – это
тангенциальная сила, приходящаяся на единицу поверхности,
которая также называется напряжением сдвига t , выраженная в
паскалях (Па), которую необходимо приложить для того, чтобы
переместить слой жидкости площадью 1 м2 со скоростью (v) 1
метр в секунду (м.с-1), находящийся на расстоянии (х) 1 метр
относительно другого слоя, параллельно площади скольжения.
37
38. Вязкость (капиллярный метод)
Методика. Испытуемую жидкость,имеющую температуру 20оС, если в
частной статье не обозначена другая
температура, заливают в вискозиметр
через трубку (L) в таком количестве, чтобы
заполнить расширение (А), но при этом
уровень жидкости в расширении (В) должен
остаться ниже выхода к вентиляционной
трубке (М). Вискозиметр в вертикальном
положении погружают в водяную баню при
температуре (20+/-0,1)оС, если в частной
статье не указана другая температура,
удерживая его в этом положении не менее
30 минут для установления температурного
равновесия. Трубку (М) закрывают и
повышают уровень жидкости в трубке (N)
таким образом, чтобы она находилась
примерно на 8 мм выше метки (Е).
Удерживают жидкость на этом уровне,
закрыв трубку (N) и открыв трубку (М).
Затем открывают трубку (N) и измеряют
время, за которое уровень жидкости
снизится от метки (Е) до метки (F),
секундомером с точностью до одной пятой
секунды.
38
39. Температурные пределы перегонки
3940. Температура плавления
1. Капиллярный метод определения температурыплавления. Температура плавления, определенная
капиллярным методом, представляет собой температуру, при
которой последняя твердая частичка уплотненного столбика
вещества в капиллярной трубке переходит в жидкую фазу.
2. Открытый капиллярный метод - применяют для
веществ, имеющих аморфную структуру, не растирающихся в
порошок и плавящихся ниже температуры кипения воды,
таких как жиры, воск, парафин, вазелин, смолы.
3. Метод мгновенного плавления - применяют для твердых
веществ, легко превращаемых в порошок.
4. Температура каплепадения - температура, при которой в
условиях, приведенных ниже, первая капля расплавленного
испытуемого вещества падает из чашечки (жиры, воски,
масла).
5. Температура затвердевания – максимальная температура,
при которой происходит затвердевание переохлажденной жидкости.
40
41. Определение температуры плавления (инструментальное)
Видео процесса плавленияЦветное видео высокого разрешения позволяет изучать
вещества, которые плавятся с разложением или имеют
окраску. С помощью приборов можно также изучать явления
41
термохромизма.
42. Подлинность (методы)
1. Химические реакции подлинности:А. Общие реакции на подлинность по
функциональным группам (первичные
ароматические амины, алкалоиды,
сложные эфиры и др.)
Б. Специфичные реакции на ионы
В. Специфичные реакции на
органические вещества
42
43. Примеры реакций идентификации по функциональным группам
Реакция на первичную ароматическую аминогруппу:43
44. Примеры реакций идентификации по функциональным группам
Реакция на первичную аминогруппу(нингидриновая реакция):
44
45. Специфические реакции на ионы
4546. Специфические реакции на ионы
4647. Специфические реакции на ионы
Специфические реакции на ионыподразделяются:
1. Реакции осаждения
2. ОВ реакции
3. Реакции разложения
4. Реакции комплексообразования
47
48. Специфические реакции подлинности
4849.
4950.
5051.
5152.
5253.
5354.
5455.
5556.
5657. Подлинность (методы)
2. Инструментальные методы2.1. ИК-спектроскопия (ИК-Фурье)
2.2. Абсорционная спектрофотометрия
в УФ и/или видимой области спектра
2.3. Хроматографические методы (ТСХ,
ГХ, ЖХ)
2.4. Электрофорез, капиллярный
электрофорез (включая пептидное
картирование)
57
58. Подлинность (методы)
3. Физические методы (определениефизических констант):
3.1. Температура плавления, кипения,
температурные пределы перегонки.
3.2. Относительная плотность.
3.3. Показатель преломления.
3.4. Угол оптического вращения.
3.5. Определение вязкости.
58
59. Подлинность (доказательство)
Установление подлинности ЛВ проводитсякак минимум 2 методами!
Первая идентификация – специфичный
инструментальный метод (как правило ИКспектрометрия) + дополнительныйметод
(например, хроматографический или
химический метод)
Вторая идентификация – подтверждение
подлинности (используются определение
физических констант, дополнительных
химических методов, абсорбционная
спектрофотометрия и др.).
59
60. Примеси (классификация)
1. Общие технологические примеси – попадающие в процессепроизводства.
1.1. Реагентные примеси (SO42-,Cl-, сульфатная зола и др.)
1.2. Примеси от контакта с технологическим оборудованием (HM,
As, Pb, Cd, Fe и др.)
1.3. Остаточные органические растворители
1.4. Вода, влага
2. Специфические примеси – характерны для конкретного ЛВ и
включают:
2.1. Полупродукты синтеза и специфические реагенты
2.2. Побочные продукты синтеза
2.3. Сопутствующие примеси (химически родственные аналоги и
остаточные кол-ва пестицидов и супертоксикантов – для ЛВ
природного происхождения)
2.4. Стереоизомеры-примеси (примеси энантиомеров)
2.5. Продукты разложения и взаимодействия с технологическими
примесями, влагой, кислородом воздуха, органическими
растворителями и др.
3. Механические примеси
60
61. Примеси
1. Летучие (характеризуются потерей в массе привысушивании).
2. Неорганические (устанавливаются при определении
сульфатной золы, тяжелых металлов и т.д.).
3. Родственные по структуре примеси (определяются
хроматографическими методами или электрофорезом).
Отдельно классифицируют токсичные
(оказывают влияние на фармакологический
эффект – т.е. являются недопустимыми) и
нетоксичные (указывают на степень очистки
ЛВ) примеси.
61
62. Потеря в массе при высушивании (метод гравиметрии)
Является суммарным неспецифичным показателем,характеризующим наличие воды (влаги), остаточных 62
органических растворителей в ЛВ
63. Определение воды
1. Дистилляция (отгонка) – для жидкостей2. Титриметрический метод (метод К.
Фишера, микрометод) – для твердых веществ
63
64. Физические и химические свойства, характеризующие чистоту
Прозрачность и степень мутности. Прозрачные растворы –при освещении их электролампой на черном фоне не
наблюдается присутствие нерастворенных частиц. Степень
мутности устанавливают путем сравнения испытуемого
вещества с эталоном (или с растворителем).
Окраску жидкостей устанавливают путем сравнения
испытуемых растворов с равным объем одного из эталонов при
дневном освещении на матово-белом фоне.
Адсорбционная способность – устанавливается по
обесцвечиванию красителя (метиленовый синий) в растворе ЛВ
определенной концентрации.
Примеси окрашенных веществ (светопоглощающие примеси)
– для неокрашенных веществ определяется абсорбция
раствора ЛВ в воде или органическом растворителе в видимой
области спектра.
64
65. Определение золы
Метод гравиметрии1. Общая зола (ЛРС, ряд органических
ЛВ) – сжигание навески (1.0000 г)
испытуемого образца в тигле при Т
около 500оС (30 мин), после
охлаждения определяют массу остатка.
2. Сульфатная зола - навеску
смачивают 1 мл Н2SO4 и далее
поступают как при определении общей
золы.
65
66. Определение «тяжелых» металлов
А. Стадия пробоподготовки:1. Растворение в воде (для ЛВ, хорошо растворимых в воде) или
в смеси с органическими растворителями (ацетон, диоксан);
2. «Мокрая» минерализация (для органических веществ) –
2.1. сжигание ЛВ со смесью MgSO4 и H2SO4 (Т=800оС).
2.2. минерализация смесью H2SO4 и HNO3 (нагревание до
200оC).
2.3. минерализация с использованием СВЧ-нагревания
(тефлоновые сосуды, 2,5 ГГц).
3. «Сухая» минерализация – сплавление с MgO (Т=600оС).
Б. Качественный и/или полуколичественный анализ
(химическая реакция с сульфид-ионом):
1. Качественный – безэталонный (отсутствие окраски с
реагентом)
2. Полуколичественный анализ – сравнение окраски с эталоном,
содержащим предельное количество ионов свинца (эталона).
66
В. Количественный анализ – метод ААС или АЭС.
67. Остаточные органические растворители (классификация)
В основе классификации лежит потенциальнаяопасность растворителей для организма человека и
окружающей среды.
Класс 1. Растворители, использования которых
следует избегать (канцерогенные вещества и
супертоксиканты окружающей среды – бензол, ТХУ,
1,2-дихлорэтан, 1,1-дихлорэтен, 1,1,1-трихлорэтан).
Класс 2. Растворители, использование которых
следует ограничивать (негенотоксичные
канцерогены, вещества с существенной
токсичностью) – ацетонитрил, гексан, диоксан,
ксилол, метанол, нитрометан, пиридин, хлороформ,
толуол, этилеггликоль и др.
67
68. Остаточные органические растворители (классификация, продолжение)
Класс 3. Малотоксичные растворители (снизким потенциалом токсичности у человека,
не требуют установления предельных
содержаний – менее 5000 ppm (мкг/г) или
0,5%) – ацетон, бутанол-1, бутанол-2, гептан,
ДМСО, пентан, уксусная кислота, пропанол-1,
пропанол-2, этанол, ТГФ, пентан и др.
Класс 4. Растворители, для которых
отсутствуют необходимые данные о
токсичности (изооктан, петролейный эфир,
трифторуксусная кислота и др.).
68
69. Остаточные органические растворители
Метод газовой хроматографии (ГХскрининг)А. Подготовка образца и раствора
сравнения
1. Растворение навески испытуемого образца
в воде (для ЛВ, растворимых в воде).
2. Растворение навески испытуемого образца
в диметилформамиде (ДМФА).
3. Растворение навески испытуемого образца
в 1,3-диметил-2-имидазолидиноне.
Поскольку большинство органических растворителей
«включены» в кристаллическую решетку (или в
структуру в виде сольватов) ЛВ, пробоподготовка
должна включать полное растворение образца с
«разрушением» решетки и возможных сольватов.
CH3
H
N
CH3
O
CH3
N
O
N
CH3
69
70. Остаточные органические растворители (анализ)
Б. Парофазовая пробоподготовка –проводится для перевода ООР из раствора в
парогазовую фазу (нагревание в герметично
укупоренном сосуде).
В. Газохроматографический анализ парогазовой фазы (полуколичественный анализ с
разделением на капиллярной колонке средней
полярности).
70
71. Специфические примеси
1. Полупродукты синтеза и специфические реагенты(включая катализаторы)
1.1. Неорганические вещества – катионы, анионы,
комплексные соединения
1.2. Органические вещества
1.3. Генетически-модифицированные микроорганизмы,
вирусы и др.
O
N
N
HN
N
N
N
CH3
Ирбесартан (примесь азид-иона)
71
72. Специфические примеси
Наибольшая группа примесей в органических ЛВ –родственные по химической структуре химические
вещества (число их ограничено пока только
возможностями методов разделения и детекции). Чем
сложнее хим. структура – тем большее количество
примесей необходимо нормировать.
O
H3C
H3C
CH3
O
H
H
CH3
H
O
H
H3C
O
O
CH3
O
H
H
S
O
H
O
S
H
H
Br
O
H
CH3
O
CH3
H
O
S
H
O
O
H3C
CH3
CH3
Спиронолактон
H3C
O
H
H
O
CH3
H3C
O
CH3
H
H
H
O
O
H
H
H
H
O
72
O
73. Специфические примеси
OHOH
O
Парацетамол
O2N
H3C
N
H
OH
HO
H2N
O
Побочные
продукты
синтеза
Cl
H3C
O
N
H
OH
O
H3C
H3C
N
H
Промежуточные
продукты
синтеза
N
H
Cl
OH
O
H3C
N
H
73
74. Специфические примеси
Сопутствующие примеси в ЛВ природногопроисхождения:
А. химически родственные аналоги
(обладают биологической (фармакологической)
активностью, могут быть потенциально опасны
для организма)
Б. остаточные кол-ва пестицидов и
супертоксикантов (полихлордиоксины,
полихлорбифенилы), продукты
жизнедеятельности микроорганизмов
(афлатоксины) – безусловные токсические
вещества, жестко нормируемые на уровне ppm и
ppb (мкг/г или нг/г)
74
75. Сопутствующие примеси в ЛВ природного происхождения (пример)
OHO
OH
OH
O
H
H
H
HO
H
OH
H
OH
cholic acid
H
HO
O
H
OH
ursodeoxycholic acid
H
Урсодезоксихолевая кислота
(выделяется из медвежьей желчи)
H
H
OH
OH
chenodeoxycholic acid
75
76. Специфические примеси
Продукты разложения и взаимодействия:1. с технологическими примесями (тяжелыми металлами
(d-элементы являются катализаторами многих ОВреакций, в том числе с участием O2), ионами железа,
остатками реагентов с реакционоспособными
функциональными группами),
2. с влагой (возможны реакции гидролиза (сложные
эфиры, амиды, карбаматы и др.), поглощение влаги
всегда связано с уменьшением содержания активного
вещества),
3. с кислородом воздуха (кислородочувстивительные
вещества, например, полиненасыщенные жирные
кислоты, сильные восстановители),
4. с остаточными органическими растворителями (ряд
органических растворителей – этиленоксид, дихлорметан,
дихлорэтан, уксусная кислота и др. – достаточно
реакционоспособны и реагируют с ЛВ при хранении).
76
77. Стрессовые испытания -
Стрессовые испытания Испытания устойчивости ЛВ подвоздействием ряда факторов
(температура, реагенты, освещение) с
целью доказательства селективности
методов оценки примесей, изучения
образования и идентификации
примесей, дополнительного изучения
стабильности ЛВ при хранении.
77
78. Стрессовые испытания (условия)
1. Температура – последовательноеповышение температуры при хранении
образца ЛВ на 10оС (50, 60 и т.д.);
2. Влажность (повышение отн. влажности
воздуха при хранении образца ЛВ до 75% и
выше).
3. Реагенты – растворы кислот (1М HCl),
щелочей (1М или 0,1М NaOH), H2O2 (3-30%)
при нагревании.
4. Воздействие света (УФ-свет,
интенсивность - не менее 200 Вт.ч/м2)
78
79. Количественное определение
Методы анализа (классификация,краткая характеристика, применение
для анализа ЛВ и ЛС, сравнительная
оценка) – это тема следующих как
минимум 3 лекций!
Благодарю за внимание!
4.2 Оптические методы
К этой группе относятся методы, основанные на определении показателя преломления луча света в растворе испытуемого вещества (рефрактометрия), измерении интерференции света (интерферометрия), способности раствора вещества вращать плоскость поляризованного луча (поляриметрия).
Оптические методы находят все более широкое применение в практике внутриаптечного контроля ввиду экспрессности, минимального расхода анализируемых лекарств.
Рефрактометрия использована для испытания подлинности лекарственных веществ, представляющих собой жидкости (диэтиламид никотиновой кислоты, метилсалицилат, токоферола ацетат), а во внутриаптечном контроле -- для анализа лекарственных форм, в том числе двойных и тройных смесей. Применяют также объемно-рефрактометрический анализ и рефрактометрический анализ методом полной и неполной экстракции.
Разработаны различные варианты методик анализа интерферометрическим методом лекарственных препаратов, титрованных растворов, дистиллированной воды.
Поляриметрию применяют для испытания подлинности лекарственных веществ, в молекулах которых имеется асимметрический атом углерода. Среди них большинство препаратов из групп алкалоидов, гормонов, витаминов, антибиотиков, терпенов.
В аналитической химии и фармацевтическом анализе используются рентгенорефрактометрия порошков, спектрополяриметрический анализ, лазерная интерферометрия, дисперсия вращения и круговой дихроизм.
Помимо указанных оптических методов для идентификации индивидуальных лекарственных веществ в фармацевтическом и токсикологическом анализе не теряет своего значения химическая микроскопия. Перспективно применение электронной микроскопии, особенно в фитохимическом анализе. В отличие от оптической микроскопии объект подвергается воздействию пучка электронов высоких энергий. Изображение, образованное рассеянными электронами, наблюдают на флуоресцирующем экране.
Одним из перспективных экспрессных физических методов является рентгенографический анализ. Он позволяет идентифицировать лекарственные вещества в кристаллической форме и различать при этом их полиморфное состояние. Для анализа кристаллических лекарственных веществ могут быть также применены различные виды микроскопии и такие методы, как оже-спектрометрия, фотоакустическая спектроскопия, компьютерная томография, измерения радиоактивности и др.
Эффективным недеструктивным методом является отражательная инфракрасная спектроскопия, которая используется для определения примесей различных продуктов разложения и воды, а также в анализе многокомпонентных смесей.
4.3 Абсорбционные методы
Абсорбционные методы основаны на свойствах веществ поглощать свет в различных областях спектра.
Атомно-абсорбционная спектрофотометрия основана на использовании ультрафиолетового или видимого излучения резонансной частоты. Поглощение излучения вызывается переходом электронов с внешних орбиталей атомов на орбитали с более высокой энергией. Объектами, поглощающими излучение, являются газообразные атомы, а также некоторые органические вещества. Сущность определений методом атомно-абсорбционной спектрометрии состоит в том, что через пламя, в котором распыляется анализируемый раствор пробы, проходит резонансное излучение от лампы с полым катодом. Это излучение попадает на входную щель монохроматора, причем из спектра выделяется только резонансная линия испытуемого элемента. Фотоэлектрическим методом измеряют уменьшение интенсивности резонансной линии, происходящей вследствие поглощения ее атомами определяемого элемента. Расчет концентрации производят с помощью уравнения, отражающего ее зависимость от ослабления интенсивности излучения источника света, длины поглощающего слоя и коэффициента поглощения света в центре линии поглощения. Метод отличается высокой избирательностью и чувствительностью.
Поглощение резонансных линий измеряют на атомно-абсорбцион- ных спектрофотометрах типа "Спектр-1", "Сатурн" и др. Точность определений не превышает 4%, предел обнаружения достигает 0,001 мкг/мл. Это свидетельствует о высокой чувствительности метода. Он находит все более широкое применение для оценки чистоты лекарственных препаратов, в частности определения минимальных примесей тяжелых металлов. Перспективно использование атомно-абсорбционной спектрофотометрии для анализа поливитаминных препаратов, аминокислот, барбитуратов, некоторых антибиотиков, алкалоидов, галогенсодержащих лекарственных веществ, ртутьсодержащих соединений.
Возможно также применение в фармации рентгеновской абсорбционной спектроскопии, основанной на поглощении атомами рентгеновского излучения.
Ультрафиолетовая спектрофотометрия -- наиболее простой и широко применяемый в фармации абсорбционный метод анализа. Его используют на всех этапах фармацевтического анализа лекарственных препаратов (испытания подлинности, чистоты, количественное определение). Разработано большое число способов качественного и количественного анализа лекарственных форм методом ультрафиолетовой спектрофотометрии. Для идентификации могут быть использованы атласы спектров лекарственных веществ, систематизирующие сведения о характере спектральных кривых и значениях удельных показателей поглощения.
Известны различные варианты использования метода УФ-спектрофотометрии для идентификации. При испытаниях на подлинность идентифицируют лекарственные вещества по положению максимума светопоглощения. Чаще в фармакопейных статьях приведены положения максимума (или минимума) и соответствующие им значения оптических плотностей. Иногда используют метод, основанный на вычислении отношения оптических плотностей при двух длинах волн (они обычно соответствуют двум максимумам или максимуму и минимуму светопоглощения). Идентифицируют целый ряд лекарственных веществ также по удельному показателю поглощения раствора.
Весьма перспективно для идентификации лекарственных веществ использование таких оптических характеристик, как положение полосы поглощения в шкале длин волн, частота в максимуме поглощения, значение пиковой и интегральной интенсивности, полуширина и асимметрия полос, сила осциллятора. Эти параметры делают более надежной идентификацию веществ, чем установление длины волны максимума светопоглощения и удельного показателя поглощения. Эти константы, позволяющие охарактеризовать наличие связи между УФ-спектром и структурой молекулы, были установлены и использованы для оценки качества лекарственных веществ, содержащих гетероатом кислорода в молекуле (В.П.Буряк).
Объективный выбор оптимальных условий количественного спектрофотометрического анализа можно осуществить только предварительным исследованием констант ионизации, влияния природы растворителей, рН среды и других факторов на характер спектра поглощения.
В НТД приведены различные способы использования УФ-спектрофотометрии для количественного определения лекарственных веществ, являющихся витаминами (ретинола ацетат, рутин, цианокобаламин), стероидными гормонами (кортизона ацетат, преднизон, прегнин, тестостерона пропионат), антибиотиками (натриевые соли оксациллина и метициллина, феноксиметилпенциллин, левомицетина стеарат, гризеофульвин). В качестве растворителей для спектрофотометрических измерений обычно используют воду или этанол. Расчет концентрации проводят различными способами: по стандарту, удельному показателю поглощения или калибровочному графику.
Количественный спектрофотометрический анализ целесообразно комбинировать с установлением подлинности по УФ-спектру. В этом случае раствор, приготовленный из одной навески, можно использовать для обоих этих испытаний. Чаще всего при спектрофотометрических определениях применяют способ, основанный на сравнении оптических плотностей анализируемого и стандартного растворов. Определенных условий анализа требуют лекарственные вещества, способные образовывать кислотно-основные формы в зависимости от рН среды. В таких случаях необходимо предварительно подбирать условия, в которых вещество в растворе полностью будет находиться в одной из таких форм.
Для уменьшения относительной погрешности фотометрического анализа, в частности снижения систематической ошибки, весьма перспективно использование стандартных образцов лекарственных веществ. Учитывая сложность получения и высокую стоимость, они могут быть заменены эталонами, приготавливаемыми из доступных неорганических соединений (дихромата калия, хромата калия).
В ГФ XI расширена область применения УФ-спектрофотометрии. Метод рекомендован для анализа многокомпонентных систем, а также для анализа лекарственных веществ, которые сами не поглощают свет в ультрафиолетовой и видимой областях спектра, но могут быть превращены в поглощающие свет соединения с помощью различных химических реакций.
Дифференциальные методы позволяют расширить область применения фотометрии в фармацевтическом анализе. Они дают возможность повысить ее объективность и точность, а также анализировать высокие концентрации веществ. Кроме того, этими методами можно анализировать многокомпонентные смеси без предварительного разделения.
Метод дифференциальной спектрофотометрии и фотоколориметрии включен в ГФ XI, вып. 1 (с. 40). Сущность его заключается в измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество испытуемого вещества. Это приводит к изменению рабочей области шкалы прибора и снижению относительной погрешности анализа до 0,5--1%, т.е. такой же, как и у титриметрических методов. Хорошие результаты были получены при использовании вместо растворов сравнения нейтральных светофильтров с известной оптической плотностью; входящих в комплект спектрофотометров и фотоколориметров (В.Г.Беликов).
Дифференциальный метод нашел применение не только в спектрофотометрии и фотоколориметрии, но и в фототурбидиметрии, фотонефелометрии, интерферометрии. Дифференциальные методы могут быть распространены и на другие физико-химические методы. Большие перспективы для анализа лекарств имеют и методы химического дифференциального анализа, основанные на использовании таких химических воздействий на состояние лекарственного вещества в растворе, как изменение рН среды, смена растворителя, изменение температуры, влияние электрических, магнитных, ультразвуковых полей и др.
Широкие возможности открывает в количественном спектрофотометрическом анализе один из вариантов дифференциальной спектрофотометрии -- ?Е-метод. Он основан на превращении анализируемого вещества в таутомерную (или иную) форму, отличающуюся по характеру светопоглощения.
Новые возможности в области идентификации и количественного определения органических веществ открывает использование производной УФ-спектрофотометрии. Метод основан на выделении индивидуальных полос из УФ-спектров, представляющих собой сумму налагающихся полос поглощения или полос, не имеющих четко выраженного максимума поглощения.
Производная спектрофотометрия дает возможность идентификации сходных по химической структуре лекарственных веществ или их смесей. Для повышения избирательности качественного спектрофотометрического анализа применяют способ построения вторых производных УФ-спектров. Вторую производную можно рассчитать способом численного дифференцирования.
Разработан унифицированный метод получения производных от спектров поглощения, который учитывает особенности характера спектра. Показано, что вторая производная имеет разрешающую способность примерно в 1,3 раза больше по сравнению с непосредственной спектрофотометрией. Это позволило использовать данный метод для идентификации кофеина, теобромина, теофиллина, папаверина гидрохлорида и дибазола в лекарственных формах. Вторая и четвертая производные в количественном анализе более эффективны по сравнению с титриметрическими методами. Продолжительность определения сокращается в 3-4 раза. Определение указанных препаратов в смесях оказалось возможным вне зависимости от характера поглощения сопутствующих веществ или при существенном уменьшении влияния их светопоглощения. Это позволяет исключить трудоемкие операции по разделению смесей.
Использование в спектрофотометрическом анализе комбинированного полинома позволило исключить влияние нелинейного фона и разработать методики количественного определения ряда препаратов в лекарственных формах, не требующие сложных расчетов результатов анализа. Комбинированный полином успешно применен при изучении процессов, происходящих при хранении лекарственных веществ и в химико-токсикологических исследованиях, так как позволяет уменьшить влияние светопоглощающих примесей (Е.Н.Вергейчик).
Спектроскопия комбинационного рассеяния (СКР) отличается от других спектроскопических методов по чувствительности, большому выбору растворителей и интервалов температур. Наличие отечественного КР-спектрометра марки ДСФ-24 позволяет применять этот метод не только для установления химической структуры, но и в фармацевтическом анализе.
Не получил еще должного развития в практике фармацевтического анализа метод спектрофотометрического титрования. Этот метод дает возможность выполнения безындикаторного титрования многокомпонентных смесей с близкими значениями рК на основе последовательного изменения оптической плотности в процессе титрования в зависимости от объема добавляемого титранта.
Фотоколориметрический метод широко применяется в фармацевтическом анализе. Количественное определение этим методом в отличие от УФ-спбктрофотометрии осуществляют в видимой области спектра. Определяемое вещество с помощью какого-либо реагента переводят в окрашенное соединение, а затем измеряют интенсивность окраски раствора на фотоколориметре. Точность определений зависит от выбора оптимальных условий протекания химической реакции.
Очень широко в фотометрическом анализе используются методики анализа препаратов, производных первичных ароматических аминов, основанные на использовании реакций диазотирования и азосочетания. В качестве азосоставляющего широко применяют N -(1-нафтил)-этилендиамин. Реакция образования азокрасителей лежит в основе фотометрического определения многих препаратов, производных фенолов.
Фотоколориметрический метод включен в НТД для количественного определения ряда нитропроизводных (нитроглицерин, фурадонин, фуразолидон), а также препаратов витаминов (рибофлавин,фолиевая кислота) и сердечных гликозидов (целанид). Разработаны многочисленные методики фотоколориметрического определения препаратов в лекарственных формах. Известны различные модификации фотоколориметрии и способы расчета концентрации в фотоколориметрическом анализе.
Перспективными для применения в качестве цветореагентов в фотометрическом анализе оказались такие поликарбонильные соединения, как биндон (ангидро-бис-индандион-1,3), аллоксан (тетраоксогекса-гидропиримидин), натриевая соль 2-карбэтоксииндандиона-1,3 и некоторые ее производные. Установлены оптимальные условия и разработаны унифицированные способы идентификации и спектрофотометрического определения в видимой области лекарственных веществ, содержащих первичную ароматическую или алифатическую аминогруппу, остаток сульфонил мочевины или являющимися азотсодержащими органическими основаниями и их солями (В.В.Петренко).
Широко используют в фотоколориметрии реакции окрашивания, основанные на образовании полиметиновых красителей, которые получаются при разрыве пиридинового или фуранового циклов либо при некоторых реакциях конденсации с первичными ароматическими аминами (А.С.Бейсенбеков).
Для идентификации и спектрофотометрического определения в видимой области спектра лекарственных веществ, производных ароматических аминов, тиолов, тиоамидов и других меркаптосоединений использованы в качестве цветореагентов N -хлор-, N -бензолсульфонил- и N -бензолсульфонил-2-хлор-1,4-бензохинонимина.
Один из вариантов унификации способов фотометрического анализа основан на косвенном определении по остатку нитрита натрия, вводимого в реакционную смесь в виде стандартного раствора, взятого в избытке. Избыток нитрита определяют затем фотометрически реакцией диазотирования с помощью этакридина лактата. Такой прием применен для косвенного фотометрического определения азотсодержащих лекарственных веществ по нитрит-иону, образующемуся в результате их превращений (гидролиза, термического разложения). Унифицированная методика позволяет осуществлять контроль качества более 30 таких лекарственных веществ в многочисленных лекарственных формах (П.Н.Ивахненко).
Фототурбидиметрия и фотонефелометрия - это методы, имеющие большие возможности, но пока ограниченно применяющиеся в фармацевтическом анализе. Основаны на измерении света, поглощенного (турбидиметрия) или рассеянного (нефелометрия) взвешенными частицами анализируемого вещества. С каждым годом методы совершенствуются. Рекомендуют, например, хронофототурбидиметрию в анализе лекарственных веществ. Сущность метода заключается в установлении изменений светопогашений во времени. Описано также применение термонефелометрии, основанной на установлении зависимости концентрации вещества от температуры, при которой наступает помутнение раствора препарата.
Систематические исследования в области фототурбидиметрии, хронофототурбидиметрии и фототурбидиметрического титрования показали возможность применения фосфорно-вольфрамовой кислоты для количественного определения азотсодержащих лекарственных веществ. В фототурбидиметрическом анализе использован как непосредственный, так и дифференциальный метод, а также автоматическое фототурбидиметрическое титрование и хронофототурбидиметрическое определение двухкомпонентных лекарственных форм (А.И.Сичко).
Инфракрасная (ИК) спектроскопия характеризуется широкой информативностью, что создает возможность объективной оценки подлинности и количественного определения лекарственных веществ. ИК-спектр однозначно характеризует всю структуру молекулы. Различия в химическом строении меняют характер ИК-спектра. Важные преимущества ИК-спектрофотометрии -- специфичность, быстрота выполнения анализа, высокая чувствительность, объективность получаемых результатов, возможность анализа вещества в кристаллическом состоянии.
ИК-спектры измеряют, используя обычно взвеси лекарственных веществ в вазелиновом масле, собственное поглощение которого не мешает идентификации анализируемого соединения. Для установления подлинности используют, как правило, расположенную в интервале частот от 650 до 1800 см -1 так называемую область "отпечатков пальцев" (650--1500 см -1), а также валентные колебания химических связей
С=0, С=С, С=N
В ГФ XI рекомендованы два способа установления подлинности лекарственных веществ но ИК-спектрам. Один из них основан на сравнении ИК-спектров испытуемого вещества и его стандартного образца. Спектры должны быть сняты в идентичных условиях, т.е. образцы должны быть в одинаковом агрегатном состоянии, в одной и той же концентрации, единой должна быть скорость регистрации и т.д. Второй способ заключается в сравнении ИК-спектра испытуемого вещества с его стандартным спектром. В этом случае необходимо строго соблюдать условия, предусмотренные для снятия стандартного спектра, приведенные в соответствующей НТД (ГФ, ВФС, ФС). Полное совпадение полос поглощения свидетельствует об идентичности веществ. Однако полиморфные модификации могут давать различные ИК-спектры. В таком случае для подтверждения идентичности необходимо перекристаллизовать испытуемые вещества из одного и того же растворителя и вновь снять спектры.
Подтверждением подлинности лекарственного вещества может служить также интенсивность поглощения. Для этой цели используют такие константы как показатель поглощения или величина интегральной интенсивности поглощения, равная площади, которую огибает кривая на спектре поглощения.
Установлена возможность использования ИК-спектроскопии для идентификации большой группы лекарственных веществ, содержащих в молекуле карбонильные группы. Подлинность устанавливают по характеристическим полосам поглощения в следующих областях: 1720-1760, 1424-1418, 950-в00 см -1 для карбоновых кислот; 1596-1582, 1430-1400, 1630-1612, 1528-1518 см -1 для аминокислот; 1690--1670, 1615--1580 см -1 для амидов; 1770--1670 см -1 для производных барбитуровой кислоты; 1384--1370, 1742--1740, 1050 см -1 для терпеноидов; 1680--1540, 1380--1278 см -1 для антибиотиков тетрациклинового ряда; 3580-3100, 3050-2870, 1742-1630, 903-390 см -1 для стероидов (А.Ф.Мынка).
Метод ИК-спектроскопии включен в фармакопеи многих зарубежных стран и в МФ III, где использован для идентификации более 40 лекарственных веществ. Методом ИК-спектрофотометрии можно проводить не только количественную оценку лекарственных веществ, но и исследование таких химических превращений, как диссоциация, сольволиз, метаболизм, полиморфизм и т.д.
4.4 Методы, основанные на испускании излучения
К этой группе методов относят фотометрию пламени, флуоресцентные и радиохимические методы.
В ГФ XI включена эмиссионная и пламенная спектрометрия для целей качественного и количественного определения химических элементов и их примесей в лекарственных веществах. Измерение интенсивности излучения спектральных линий испытуемых элементов выполняют на отечественных пламенных фотометрах ПФЛ-1, ПФМ, ПАЖ-1. Регистрирующими системами служат фотоэлементы, связанные с цифровыми и печатающими устройствами. Точность определений методами эмиссионной, как и атомно-абсорбционной, пламенной спектрометрии находится в пределах 1--4%, предел обнаружения может достигать 0,001 мкг/мл.
Количественное определение элементов методом эмиссионной пламенной спектрометрии (пламенной фотометрии) основано на установлении зависимости между интенсивностью спектральной линии и концентрацией элемента в растворе. Сущность выполнения испытания состоит в распылении анализируемого раствора до состояния аэрозоля в пламени горелки. Под воздействием температуры пламени происходят испарение растворителя и твердых частиц из капель аэрозоля, диссоциация молекул, возбуждение атомов и возникновение их характеристического излучения. С помощью светофильтра или монохроматора излучение анализируемого элемента отделяется от других и, попадая на фотоэлемент, вызывает фототок, который измеряется с помощью гальванометра или потенциометра.
Пламенная фотометрия использована для количественного анализа натрий-, калий- и кальций-содержащих препаратов в лекарственных формах. На основе исследования влияния на эмиссию определяемых катионов, органических анионов, вспомогательных и сопутствующих компонентов были разработаны методики количественного определения натрия гидрокарбоната, натрия салицилата, ПАСК-натрия, билигноста, гексенала, натрия нуклеината, кальция хлорида и глюконата, бепаска и др. Предложены методики одновременного определения двух солей с разными катионами в лекарственных формах, например калия иодида -- натрия гидрокарбоната, кальция хлорида -- калия бромида, калия иодида -- натрия салицилата и др.
Люминесцентные методы основаны на измерении вторичного излучения, возникающего в результате воздействия света на анализируемое вещество. К их числу относят флуоресцентные методы, хемилюминесценцию, рентгенофлуоресценцию и др.
Флуоресцентные методы основаны на способности веществ флуоресцировать в УФ-свете. Эта способность обусловлена структурой либо самих органических соединений, либо продуктов их диссоциации, сольволиза и других превращений, вызванных воздействием различных реактивов.
Флуоресцирующими свойствами обладают обычно органические соединения с симметричной структурой молекул, в которых имеются сопряженные связи, нитро-, нитрозо-, азо-, амидо-, карбоксильная или карбонильная группы. Интенсивность флуоресценции зависит от химической структуры и концентрации вещества, а также других факторов.
Флуориметрия может быть использована как для качественного, так и для количественного анализа. Количественный анализ выполняют на спектрофлуориметрах. Принцип их работы состоит в том, что свет от ртутно-кварцевой лампы через первичный светофильтр и конденсор падает на кювету с раствором испытуемого вещества. Расчет концентрации проводят по шкале стандартных образцов флуоресцирующего вещества известной концентрации.
Разработаны унифицированные методики количественного спект- рофлуориметрического определения производных п-аминобензолсульфамида (стрептоцид, сульфацил-натрий, сульгин, уросульфан и др.) и п-аминобензойной кислоты (анестезин, новокаин, новокаинамид). Водно-щелочные растворы сульфаниламидов имеют наибольшую флуоресценцию при рН б--8 и 10--12. Кроме того, сульфаниламиды, содержащие в молекуле незамещенную первичную ароматическую аминогруппу, после нагревания с о-фталевым альдегидом в присутствии серной кислоты приобретают интенсивную флуоресценцию в области 320--540 нм. В той же области флуоресцируют производные барбитуровой кислоты (барбитал, барбитал-натрий, фенобарбитал, этаминал-натрий) в щелочной среде (рН 12--13) с максимумом флуоресценции при 400 нм. Предложены высокочувствительные и специфичные методики спектрофлуориметрического определения антибиотиков: тетрациклина, окситетрациклина гидрохлорида, стрептомицина сульфата, пассомицина, флоримицина сульфата, гризеофульвина и сердечного гликозида целанида (Ф.В.Бабилев). Проведены исследования спектров флуоресценции ряда лекарственных средств, содержащих природные соединения: производные кумарина, антрахинона, флавоноидов (В.П.Георгиевский).
Выявлены комплексообразующие группировки у 120 лекарственных веществ, производных оксибензойной, оксинафтойной, антраниловой кислот, 8-оксихинолина, оксипиридина, 3- и 5-оксифлавона, птеридина и др. Указанные группировки способны образовывать флуоресцирующие комплексы с катионами магния, алюминия, бора, цинка, скандия при возбуждении флуоресценции от 330 нм и выше и ее излучении при длинах волн, превышающих 400 нм. Проведенные исследования позволили разработать методики флуориметрирования 85 лекарственных средств (А.А.Хабаров).
Наряду с производной спектрофотометрией в фармацевтическом анализе обоснована возможность применения производной спектрофлуориметрии. Спектры снимают на флуоресцентном спектрофотометре МПФ-4 с термостатирующей ячейкой, а производные находят аналогичным дифференцированием с помощью компьютера. Метод использован для разработки простых, точных и высокочувствительных методик количественного определения гидрохлоридов пиридоксина и эфедрина в лекарственных формах в присутствии продуктов разложения.
Перспективность использования рентгеновской флуоресценции для определения малых количеств примесей в лекарственных препаратах обусловливается высокой чувствительностью и возможностью выполнения анализа без предварительного разрушения вещества. Метод рентгенофлуоресцентной спектрометрии оказался перспективным для количественного анализа веществ, имеющих в молекуле такие гетероатомы, как железо, кобальт, бром, серебро и др. Принцип метода заключается в сравнении вторичного рентгеновского излучения элемента в анализируемом и стандартном образце. Рентгенофлуоресцентная спектрометрия относится к числу методов, не требующих предварительных деструктивных изменений. Выполняют анализ на отечественном спектрометре РС-5700. Продолжительность анализа 15 мин.
Хемилюминесценция -- метод, заключающийся в использовании энергии, возникающей в процессе химических реакций.
Эта энергия служит источником возбуждения. Ее излучают при окислении некоторые барбитураты (особенно фенобарбитал), гидразиды ароматических кислот и другие соединения. Это создает большие возможности использования метода для определения очень малых концентраций веществ в биологическом материале.
Радиохимические методы находят все более широкоеприменение в фармацевтическоманализе. Радиометрический анализ, основанный на измерении?- или?-излучения с помощью спектрометров, использован (наряду с другими параметрами для оценки качества фармакопейных радиоактивных препаратов. Широко применяют в различных областях техники и особенно в аналитической химии высокочувствительные методы анализа с применением радиоактивных изотопов (меченых атомов). Для обнаружения следов примесей в веществах используют активационный анализ; для определения в смесях близких по свойствам трудноразделяемых компонентов -- метод изотопного разбавления. Применяют также радиометрическое титрование и радиоактивные индикаторы. Оригинальным вариантом сочетания радиоизотопного и хроматографического методов является изучение диффузионно-осадочных хроматограмм в тонком слое желатинового геля с помощью радиоактивных индикаторов.
4.5 Методы, основанные на использовании магнитного поля
Методы ЯМР-, ПМР-спектроскопии, а также масс-спектрометрии отличаются высокой специфичностью, чувствительностью и используются для анализа многокомпонентных смесей, в том числе лекарственных форм без предварительного их разделения.
Метод спектроскопии ЯМР используют для испытания подлинности лекарственных веществ, которая может быть подтверждена либо по полному набору спектральных параметров, характеризующих структуру данного соединения, либо по наиболее характерным сигналам спектра. Подлинность можно также установить с помощью стандартного образца, добавляя определенное его количество к анализируемому раствору. Полное совпадение спектров анализируемого вещества и его смеси со стандартным образцом указывает на их идентичность.
Регистрацию ЯМР-спектров выполняют на спектрометрах с рабочими частотами 60 мГц и более, используя такие основные характеристики спектров, как химический сдвиг, мультиплетность сигнала резонанса, константу спин-спинового взаимодействия, площадь сигнала резонанса. Наиболее обширную информацию о молекулярной структуре анализируемого вещества дают спектры ЯМР 13 С и 1 Н.
Надежная идентификация препаратов гестагенных и эстрогенных гормонов, а также их синтетических аналогов: прогестерона, прегнина, этинилэстрадиола, метилэстрадиола, эстрадиола дипропионата и др. -- может быть осуществлена методом спектроскопии ЯМР 1 Н в деитерированном хлороформе на спектрометре УН-90 с рабочей частотой 90 мГц (внутренний стандарт -- тетраметилсилан).
Систематические исследования позволили установить возможность применения спектроскопии ЯМР 13 С для идентификации лекарственных веществ 10-ацилпроизводных фенотиазина (хлорацизина, фторацизина, этмозина, этацизина), 1,4-бензодиазепина (хлор-, бром- и нитропроизводные) и др. Методом спектроскопии ЯМР 1 Н и 13 С осуществлены идентификация, количественная оценка основных компонентов и примесей в препаратах и стандартных образцах природных и полусинтетических антибиотиков аминогликозидов, пенициллинов, цефалоспоринов, макролидов и др. Указанный метод использован для идентификации в унифицированных условиях ряда витаминов: липоевой и аскорбиновой кислот, липамида, холина и метилметионинсульфония хлоридов, ретинола пальмитата, кальция пантотената, эргокальциферола. Метод спектроскопии ЯМР 1 Н позволил осуществлять надежную идентификацию таких сложных по химической структуре природных соединений, как сердечные гликозиды (дигоксин, дигитоксин, целанид, дезланозид, нериолин, цимарин и др.). Для ускорения обработки спектральной информации использована ЭВМ. Ряд методик идентификации включен в ФС и ВФС (В.С.Карташов).
Количественное определение лекарственного вещества может быть также выполнено с использованием спектров ЯМР. Относительная погрешность количественных определений методом ЯМР зависит от точности измерений площадей резонансных сигналов и составляет ±2--5%. При определении относительного содержания вещества или его примеси измеряют площади сигналов резонанса испытуемого вещества и стандартного образца. Затем вычисляют количество испытуемого вещества. Для определения абсолютного содержания лекарственного вещества или примеси анализируемые образцы готовят количественно и добавляют к навеске точно отвешенную массу внутреннего стандарта. После этого выполняют регистрацию спектра, измеряют площади сигналов анализируемого вещества (примеси) и внутреннего стандарта, затем вычисляют абсолютное содержание.
Развитие импульсной техники Фурье-спектроскопии, применение ЭВМ позволили резко повысить чувствительность метода ЯМР 13 С и распространить его на количественный анализ многокомпонентных смесей биоорганических соединений, в том числе лекарственных веществ без их предварительного разделения.
Спектроскопические параметры ПМР-спектров дают целый комплекс разнообразной и весьма селективной информации, который может быть использован в фармацевтическом анализе. Следует строго соблюдать условия регистрации спектров, так как на значения химических сдвигов и другие параметры оказывают влияние тип растворителя, температура, рН раствора, концентрация вещества.
Если полная интерпретация ПМР-спектров затруднена, то выделяют только характерные сигналы, по которым идентифицируют испытуемое вещество. ПМР-спектроскопия применена для испытания подлинности многих лекарственных веществ, в том числе барбитуратов, гормональных средств, антибиотиков и др.
Поскольку метод дает информацию о наличии или отсутствии примесей к основному веществу, важное практическое значение имеет ПМР-спектроскопия для испытания лекарственных веществ на чистоту. Различия в значениях величин тех или иных констант позволяют сделать заключение о присутствии примесей продуктов разложения лекарственного вещества. Чувствительность метода к примесям колеблется в широких пределах и зависит от спектра основного вещества, наличия в молекулах тех или иных групп, содержащих протоны, растворимости в соответствующих растворителях. Минимальное содержание примеси, которое можно установить, составляет обычно 1--2%. Особенно ценной является возможность обнаружения примесей изомеров, присутствие которых невозможно подтвердить другими методами. Так, например, обнаружена примесь кислоты салициловой в кислоте ацетилсалициловой, морфина в кодеине и т.д.
Количественный анализ на основе использования ПМР-спектроскопии имеет преимущества перед другими методами, заключающиеся в том, что при анализе многокомпонентных смесей нет необходимости выделять индивидуальные компоненты для калибровки прибора. Поэтому метод широко применим для количественного анализа как индивидуальных лекарственных веществ, так и растворов, таблеток, капсул, суспензий и других лекарственных форм, содержащих один или несколько ингредиентов. Стандартное отклонение не превышает ±2,76%. Описаны способы анализа таблеток фуросемида, мепробамата, хинидина, преднизолона и др.
Расширяется диапазон применения масс-спектрометрии в анализе лекарственных веществ для идентификации и количественного анализа. Метод основан на ионизации молекул органических соединений. Он отличается большой информативностью и исключительно высокой чувствительностью. Масс-спектрометрию применяют для определения антибиотиков, витаминов, пуриновых оснований, стероидов, аминокислот и других лекарственных веществ, а также продуктов их метаболизма.
Использование лазеров в аналитических приборах значительно расширяет практическое применение УФ- и ИК-спектрофотометрии, а также флуоресцентной и масс-спектроскопии, спектроскопии комбинационного рассеяния, нефелометрии и других методов. Лазерные источники возбуждения позволяют повысить чувствительность многих методов анализа, сократить продолжительность их выполнения. Лазеры используют в дистанционном анализе в качестве детекторов в хроматографии, в биоаналитической химии и т.д.
4.6 Электрохимические методы
Эта группа методов качественного и количественного анализа основана на электрохимических явлениях, происходящих в исследуемой среде и связанных с изменениями химической структуры, физических свойств или концентрации веществ.
Потенциометрия -- метод, основанный на измерении равновесных потенциалов, возникающих на границе между испытуемым раствором и погруженным в него электродом. В ГФ XI включен метод потенциометрического титрования, заключающийся в установлении эквивалентного объема титранта путем измерения ЭДС индикаторного электрода и электрода сравнения, погруженных в анализируемый раствор. Метод прямой потенциометрии используется для определения рН (рН-метрия) и установления концентрации отдельных ионов. Потенциометрическое титрование отличается от индикаторного возможностью анализировать сильно окрашенные, коллоидные и мутные растворы, а также растворы, содержащие окислителй. Кроме того, можно последовательно оттитровать в смеси несколько компонентов в водных и неводных средах. Потенциометрический метод используют для титрования на основе реакций нейтрализации, осаждения, комплексообразования, окисления -- восстановления. Электродом сравнения во всех указанных методах служит каломельный, хлорсеребряный или стеклянный (последний не используют при анализе методом нейтрализации). Индикаторным при кислотно-основном титровании является стеклянный электрод, при комплексонометрическом -- ртутный или ион-селективный, в методе осаждения -- серебряный, в окислительно-восстановительном -- платиновый.
Измерение ЭДС, возникающей при титровании за счет разности потенциалов между индикаторным электродом и электродом сравнения, производят с помощью высокоомных рН-метров. Титрант прибавляют из бюретки равными объемами, постоянно перемешивая титруемую жидкость. Вблизи точки эквивалентности титрант прибавляют по 0,1--0,05 мл. Значение ЭДС в этой точке изменяется наиболее сильно, так как абсолютная величина отношения изменения ЭДС к приращению объема прибавляемого титранта будет при этом максимальной. Результаты титрования представляют либо графически, устанавливая точку эквивалентности на кривой титрования, либо расчетным методом. Затем вычисляют эквивалентный объем титранта по формулам (см. ГФ XI, вып. 1, с. 121).
Амперометрическое титрование с двумя индикаторными электродами, или титрование "до полного прекращения тока", основано на использовании пары идентичных инертных электродов (платина, золото), которые находятся под небольшим напряжением. Метод наиболее часто используют для нитрито- и иодометрического титрования. Точку эквивалентности находят по резкому увеличению силы тока, проходящего через ячейку (в течение 30 с) после добавления последней порции реагента. Эту точку можно установить графическим методом по зависимости силы тока от объема добавленного реагента, так же как при потенциометрическом титровании (ГФ XI, вып. 1, с. 123). Разработаны также способы биамперометрического титрования лекарственных веществ при использовании методов нитритометрии, осаждения и окисления -- восстановления.
Особенно перспективна ионометрия, использующая зависимость между ЭДС гальванической сети с ионоселективным электродом и концентрацией анализируемого иона в электродной ячейке цепи. Определения неорганических и органических (азотсодержащих) лекарственных веществ с помощью ионоселективных электродов отличаются от других методов высокой, чувствительностью, экспрессностью, хорошей воспроизводимостью результатов, несложным оборудованием, доступными реагентами, пригодностью для автоматизированного контроля и исследования механизма действия лекарств. В качестве примера можно привести способы ионометрического определения калия, натрия, галогенидов и кальцийсодержащих лекарственных веществ в таблетках и в солевых кровезамещающих жидкостях. С помощью отечественных рН-метров (рН-121, рН-673), ионометра И-115 и калий селективных электродов определяют калиевые соли различных кислот (оротовой, аспарагиновой и др.).
Полярография -- метод анализа, основанный на измерении силы тока, возникающего на микроэлектроде при электровосстановлении или электроокислении анализируемого вещества в растворе. Электролиз проводят в полярографической ячейке, которая состоит из электролизера (сосуда) и двух электродов. Один из них -- ртутный капающий микроэлектрод, а другой -- макроэлектрод, которым служит либо слой ртути на электролизере, либо внешний насыщенный каломельный электрод. Полярографический анализ может быть выполнен в водной среде, в смешанных растворителях (вода -- этанол, вода -- ацетон), в неводных средах (этаноле, ацетоне, диметилформамиде и др.). При идентичных условиях измерений для идентификации вещества используют потенциал полуволны. Количественное определение основано на измерении предельного диффузного тока испытуемого лекарственного вещества (высота волны). Для определения содержания используют метод калибровочных кривых, метод стандартных растворов и метод добавок (ГФ XI, вып. 1, с. 154). Полярографию широко используют в анализе неорганических веществ, а также алкалоидов, витаминов, гормонов, антибиотиков, сердечных гликозидов. Весьма перспективны вследствие высокой чувствительности современные методы: дифференциальная пульс-полярография, осциллографическая полярография и др.
Далеко не исчерпаны возможности электрохимических методов в фармацевтическом анализе. Разрабатываются новые варианты потенциометрии: инверсионная бестоковая хронопотенциометрия, прямая потенциометрия с помощью газового аммоний-селективного электрода и др. Расширяются исследования в области применения в фармацевтическом анализе таких методов, как кондуктометрия, основанная на исследовании электрической проводимости растворов анализируемых веществ; кулонометрия, заключающаяся в измерении количества электричества, затраченного на электрохимическое восстановление или окисление определяемых ионов.
Кулонометрия имеет ряд преимуществ перед другими физико-химическими и химическими методами. Поскольку этот метод основан на измерении количества электричества, он дает возможность непосредственно определять массу вещества, а не какое-либо свойство, пропорциональное концентрации. Вот почему кулонометрия исключает необходимость использования не только стандартных, но и титрованных растворов. Что касается кулонометрического титрования, то оно расширяет область титриметрии за счет применения различных неустойчивых электрогенерированных титрантов. Одна и та же электрохимическая ячейка может быть использована для проведения титрования с использованием различных типов химических реакций. Так, методом нейтрализации можно опредачить кислоты и основания даже в миллимолярных растворах с погрешностью не более 0,5%.
Кулонометрический метод применяют при определении малых количеств анаболических стероидов, местно-анестезирующих и других лекарственных веществ. Определению не мешают наполнители таблеток. Методики отличаются простотой, экспрессностью, быстротой и чувствительностью.
Метод диэлектрических измерений в диапазоне электромагнитных волн широко применяют для экспресс-анализа в химической технологии, пищевой промышленности и других областях. Одним из перспективных направлений является диэлькометрический контроль ферментных и других биопрепаратов. Он позволяет осуществить быструю, точную, безреагентную оценку таких параметров, как влажность, степень гомогенности и чистоты препарата. Диэлькометрический контроль является многопараметровым, испытуемые растворы могут быть непрозрачными, а измерения можно выполнять бесконтактным способом с записью результатов на ЭВМ.
4.7 Методы разделения
Из физико-химических методов разделения в фармацевтическом анализе в основном используют хроматографию, электрофорез и экстракцию.
Хроматографические методы разделения веществ основаны на их распределении между двумя фазами: подвижной и неподвижной. Подвижной фазой может быть жидкость или газ, неподвижной -- твердое вещество или жидкость, адсорбированная на твердом носителе. Относительная скорость перемещения частиц вдоль пути разделения зависит от взаимодействия их с неподвижной фазой. Это приводит к тому, что каждое из веществ проходит определенную длину пути на носителе. Отношение скорости перемещения вещества к скорости перемещения растворителя обозначают Эта величина является константой вещества для данных условий разделения и используется для идентификации.
Хроматография дает возможность наиболее эффективно осуществлять избирательное распределение компонентов анализируемого образца. Это имеет существенное значение для фармацевтического анализа, объектами исследования в котором обычно являются смеси нескольких веществ.
По механизму процесса разделения хроматографические методы классифицируют на ионообменную, адсорбционную, осадочную, распределительную, окислительно-восстановительную хроматографию. По форме проведения процесса можно выделить колоночную, капиллярную и плоскостную хроматографию. Последняя может быть выполнена на бумаге и в тонком (закрепленном или незакрепленном) слое сорбента. Хроматографические методы классифицируют также по агрегатно- му состоянию анализируемого вещества. К ним относятся различные методы газовой и жидкостной хроматографии.
Адсорбционная хроматография основана на избирательной адсорбции отдельных компонентов из раствора смеси веществ. Стационарной фазой служат такие адсорбенты, как оксид алюминия, активированный уголь и др.
Ионообменная хроматография использует ионообменные процессы, происходящие между адсорбентом и ионами электролита в анализируемом растворе. Стационарной фазой служат катион обменные или ани- онобменные смолы, содержащиеся в них ионы способны обмениваться на одноименно заряженные противоионы.
Осадочная хроматография основана на различии в растворимости веществ, образующихся при взаимодействии компонентов разделяемой смеси с осадителем.
Распределительная хроматография заключается в распределении компонентов смеси между двумя несмешивающимися жидкими фазами (подвижной и неподвижной). Стационарной фазой служит пропитанный растворителем носитель, а подвижной фазой -- органический растворитель, практически не смешивающийся с первым растворителем. При выполнении процесса в колонке происходит разделение смеси на зоны, содержащие по одному компоненту. Распределительная хроматография может выполняться также в тонком слое сорбента (тонкослойная хроматография) и на хроматографической бумаге (бумажная хроматография).
Ранее других методов разделения в фармацевтическом анализе йачали применять ионообменную хроматографию для количественного определения препаратов: солей серной, лимонной и других кислот. При этом ионообменную хроматографию сочетают с кислотно-основным титрованием. Совершенствование метода позволило, используя хроматографию ионных пар с обращенной фазой, разделять некоторые гидрофильные органические соединения. Возможно сочетание комплексонометрии с использованием катионитов в Zn 2+ -фopмe для анализа аминопроизводных в смесях и алкалоидов в экстрактах и настойках. Таким образом, сочетание ионообменной хроматографии с другими методами расширяет область ее применения.
В 1975 г. предложен новый вариант хроматографии, применяемый для определения ионов и названный ионной хроматографией. Для выполнения анализа используют колонки размером 25 Х 0,4 см. Разработана двухколоночная и одноколоночная ионная хроматография. Первая основана на ионообменном разделении ионов на одной колонке с последующим снижением фонового сигнала элюента на второй колонке и кондуктометрическим детектированием, а вторая (без подавления фонового сигнала элюента) сочетается с фотометрическим, атомно-абсорбционным и другими методами детектирования определяемых ионов.
Несмотря на ограниченное число работ по использованию ионной хроматографии в фармацевтическом анализе, очевидна перспективность этого метода для одновременного определения анионного состава многокомпонентных лекарственных форм и солевых растворов для инъекций (содержащих сульфат-, хлорид-, карбонат-, фосфат-ионы), для количественного определения гетероэлементов в органических лекарственных веществах (содержащих галогены, серу, фосфор, мышьяк), для определения уровня загрязнения воды, используемой в фармацевтической промышленности, различными анионами, для определения некоторых органических ионов в лекарственных формах.
Достоинствами ионной хроматографии являются высокая селективность определения ионов, возможность одновременного определен я органических и неорганических ионов, низкий предел обнаружена (до 10 -3 и даже 10 -6 мкг/мл), малый объем проб и простота их подготовки, быстрота выполнения анализа (за 20 мин возможно разделение до 10 ионов), простота аппаратурного обеспечения, возможность сочетания с другими аналитическими методами и расширение области применения хроматографии в отношении объектов, сходных по химической структуре и трудно разделяемых методами ТСХ, ГЖХ, ЖХВД.
Наиболее широко в фармацевтическом анализе используют хроматографию на бумаге и хроматографию в тонком слое сорбента.
В бумажной хроматографии стационарной фазой служит поверхность специальной хроматографической бумаги. Распределение веществ происходит между водой, находящейся на поверхности бумаги, и подвижной фазой. Последняя представляет собой систему, включающую несколько растворителей.
В фармацевтическом анализе при выполнении испытаний методом бумажной хроматографии руководствуются указаниями ГФ XI, вып. 1 (с. 98) и частных фармакопейных статей на соответствующие лекарственные вещества (лекарственные формы). При испытаниях подлинности хроматографируют на одном листе хроматографической бумаги одновременно испытуемое вещество и соответствующий стандартный образец. Если оба вещества идентичны, то соответствующие им пятна имеют на хроматограммах одинаковый вид и равные значения R f . Если хроматографировать смесь испытуемого вещества и стандартного образца, то при их идентичности на хроматограмме должно появляться только одно пятно. Чтобы исключить влияние условий хроматографирования на получаемые значения R f , можно пользоваться более объективной величиной R S , которая представляет собой отношение величин R f испытуемого и стандартного образцов.
При испытании на чистоту о наличии примесей судят по величине и интенсивности окраски пятен на хроматограмме. Примесь и основное вещество должны иметь разные значения R f Для полуколичественного определения содержания примеси на одном листе бумаги одновременно в одинаковых условиях получают хроматограмму испытуемого вещества, взятого в определенном количестве, и несколько хроматограмм стандартного образца, взятых в точно отмеренных количествах. Затем сравнивают между собой хроматограммы испытуемого и стандартного образцов. Заключение о количестве примеси делают по величине пятен и их интенсивности.
Подобные документы
Специфические особенности фармацевтического анализа. Испытание на подлинность лекарственных препаратов. Источники и причины недоброкачественности лекарственных веществ. Классификация и характеристика методов контроля качества лекарственных веществ.
реферат , добавлен 19.09.2010
Критерии фармацевтического анализа, общие принципы испытаний подлинности лекарственных веществ, критерии доброкачественности. Особенности экспресс-анализа лекарственных форм в условиях аптеки. Проведение экспериментального анализа таблеток анальгина.
курсовая работа , добавлен 21.08.2011
Государственное регулирование в сфере обращения лекарственных средств. Фальсификация лекарственных препаратов как важная проблем сегодняшнего фармацевтического рынка. Анализ состояния контроля качества лекарственных препаратов на современном этапе.
курсовая работа , добавлен 07.04.2016
Состояние маркетинговых исследований фармацевтического рынка ЛС. Методы анализа ассортимента лекарственных средств. Товароведческая характеристика винпоцетина. Анализ препаратов для улучшения мозгового кровообращения, разрешенных к применению в стране.
курсовая работа , добавлен 03.02.2016
Применение антибиотиков в медицине. Оценка качества, хранение и отпуск лекарственных форм. Химические строение и физико-химические свойства пенициллина, тетрациклина и стрептомицина. Основы фармацевтического анализа. Методы количественного определения.
курсовая работа , добавлен 24.05.2014
Классификация лекарственных форм и особенности их анализа. Количественные методы анализа однокомпонентных и многокомпонентных лекарственных форм. Физико-химические методы анализа без разделения компонентов смеси и после предварительного их разделения.
реферат , добавлен 16.11.2010
История развития технологии лекарственных форм и аптечного дела в России. Роль лекарств в лечении заболеваний. Правильный прием лекарственных препаратов. Способ применения и дозы. Профилактика болезней с использованием медикаментов, рекомендации врача.
презентация , добавлен 28.11.2015
Система анализа маркетинговой информации. Отбор источников информации. Анализ ассортимента аптечной организации. Характерные черты рынка лекарственных препаратов. Принципы сегментирования рынка. Основные механизмы действия противовирусных препаратов.
курсовая работа , добавлен 09.06.2013
Понятие вспомогательных веществ как фармацевтического фактора; их классификация в зависимости от происхождения и назначения. Свойства стабилизаторов, пролонгаторов и корригентов запаха. Номенклатура вспомогательных веществ в жидких лекарственных формах.
реферат , добавлен 31.05.2014
Комбинированное действие лекарственных веществ. Синергизм и его основные виды. Понятие антагонизма и антидотизма. Фармацевтическое и физико-химическое взаимодействие лекарственных средств. Основные принципы взаимодействия лекарственных веществ.
Физико-химические или инструментальные методы анализа
Физико-химические или инструментальные методы анализа основаны на измерении с помощью приборов (инструментов) физических параметров анализируемой системы, которые возникают или изменяются в ходе выполнения аналитической реакции.
Бурное развитие физико-химических методов анализа было вызвано тем, что классические методы химического анализа (гравиметрия, титриметрия) уже не могли удовлетворять многочисленные запросы химической, фармацевтической, металлургической, полупроводниковой, атомной и других отраслей промышленности, требовавших повышения чувствительности методов до 10-8 - 10-9 %, их селективности и экспрессности, что позволило бы управлять технологическими процессами по данным химического анализа, а также выполнять их в автоматическом режиме и дистанционно.
Ряд современных физико-химических методов анализа позволяют одновременно в одной и той же пробе выполнять как качественный, так и количественный анализ компонентов. Точность анализа современных физико-химических методов сопоставима с точностью классических методов, а в некоторых, например в кулонометрии, она существенно выше.
К недостаткам некоторых физико-химических методов следует отнести дороговизну используемых приборов, необходимость применения эталонов. Поэтому классические методы анализа по-прежнему не потеряли своего значения и применяются там, где нет ограничений в скорости выполнения анализа и требуется высокая его точность при высоком содержании анализируемого компонента.
Классификация физико-химических методов анализа
В основу классификации физико-химических методов анализа положена природа измеряемого физического параметра анализируемой системы, величина которого является функцией количества вещества. В соответствии с этим все физико-химические методы делятся на три большие группы:
Электрохимические;
Оптические и спектральные;
Хроматографические.
Электрохимические методы анализа основаны на измерении электрических параметров: силы тока, напряжения, равновесных электродных потенциалов, электрической проводимости, количе-ства электричества, величины которых пропорциональны содержанию вещества в анализируемом объекте.
Оптические и спектральные методы анализа основаны на измерении параметров, характеризующих эффекты взаимодействия электромагнитного излучения с веществами: интенсивности излучения возбужденных атомов, поглощения монохроматического излучения, показателя преломления света, угла вращения плоскости поляризованного луча света и др.
Все эти параметры являются функцией концентрации вещества в анализируемом объекте.
Хроматографические методы - это методы разделения однородных многокомпонентных смесей на отдельные компоненты сорбционными методами в динамических условиях. В этих условиях компоненты распределяются между двумя несмешивающимися фазами: подвижной и неподвижной. Распределение компонентов основано на различии их коэффициентов распределения между подвижной и неподвижной фазами, что при- водит к различным скоростям переноса этих компонентов из неподвижной в подвижную фазу. После разделения количественное содержание каждого из компонентов может быть определено различными методами анализа: классическими или инструментальными.
Молекулярно-абсорбционный спектральный анализ
Молекулярно-абсорбционный спектральный анализ включает в себя спектрофотометрический и фотоколориметрический виды анализа.
Спектрофотометрический анализ основан на определении спектра поглощения или измерении светопоглощения при строго определенной длине волны, которая соответствует максимуму кривой поглощения исследуемого вещества.
Фотоколориметрический анализ базируется на сравнении интенсивности окрасок исследуемого окрашенного и стандартного окрашенного растворов определенной концентрации.
Молекулы вещества обладают определенной внутренней энергией Е, составными частями которой являются:
Энергия движения электронов Еэл находящихся в электростати-ческом поле атомных ядер;
Энергия колебания ядер атомов друг относительно друга Е кол;
Энергия вращения молекулы Е вр
и математически выражается как сумма всех указанных выше энергий:
При этом, если молекула вещества поглощает излучение, то ее первоначальная энергия Е 0 повышается на величину энергии поглощенного фотона, то есть:
Из приведенного равенства следует, что чем меньше длина волны л, тем больше частота колебаний и, следовательно, больше Е, то есть энергия, сообщенная молекуле вещества при взаимодействии с электромагнитным излучением. Поэтому характер взаимодействия лучевой энергии с веществом в зависимости от длины волны света л будет различен.
Совокупность всех частот (длин волн) электромагнитного излучения называют электромагнитным спектром. Интервал длин волн разбивают на области: ультрафиолетовая (УФ) примерно 10-380 нм, видимая 380-750 нм, инфракрасная (ИК) 750-100000 нм.
Энергии, которую сообщают молекуле вещества излучения УФ- и видимой части спектра, достаточно, чтобы вызвать изменение электронного состояния молекулы.
Энергия ИК-лучей меньше, поэтому ее оказывается достаточно только для того, чтобы вызвать изменение энергии колебательных и вращательных переходов в молекуле вещества. Таким образом, в различных частях спектра можно получить различную информацию о состоянии, свойствах и строении веществ.
Законы поглощения излучения
В основе спектрофотометрических методов анализа лежат два основных закона. Первый из них - закон Бугера - Ламберта, второй закон - закон Бера. Объединенный закон Бугера - Ламберта - Бера имеет следующую формулировку:
Поглощение монохроматического света окрашенным раствором прямо пропорционально концентрации поглощающего свет вещества и толщине слоя раствора, через который он проходит.
Закон Бугера - Ламберта - Бера является основным законом светопоглощения и лежит в основе большинства фотометрических методов анализа. Математически он выражается уравнением:
Величину lg I /I 0 называют оптuческой плотностью поглощающего вещества и обозначают буквами D или А. Тогда закон можно записать так:
Отношение интенсивности потока монохроматического излучения, прошедшего через испытуемый объект, к интенсивности первоначального потока излучения называется прозрачностью, или пропусканием, раствора и обозначается буквой Т: Т = I /I 0
Это соотношение может быть выражено в процентах. Величина Т, характеризующая пропускание слоя толщиной 1 см, называется коэффициентом пропускания. Оптическая плотность D и пропускание Т связаны между собой соотношением
D и Т являются основными величинами, характеризующими поглощение раствора данного вещества с определенной его концентрацией при определенной длине волны и толщине поглощающего слоя.
Зависимость D(С) имеет прямолинейный характер, а Т(С) или Т(l) - экспоненциальный. Это строго соблюдается только для монохроматических потоков излучений.
Величина коэффициента погашения К зависит от способа выражения концентрации вещества в растворе и толщины поглощающего слоя. Если концентрация выражена в молях на литр, а толщина слоя - в сантиметрах, то он называется молярным коэффициентом погашения, обозначается символом е и равен оптической плотности раствора с концентрацией 1 моль/л, помещенного в кювету с толщиной слоя 1 см.
Величина молярного коэффициента светопоглощения зависит:
От природы растворенного вещества;
Длины волны монохроматического света;
Температуры;
Природы растворителя.
Причины несоблюдения закона Бyгера - Ламберта - Бера.
1. Закон выведен и справедлив только для монохроматического света, поэтому недостаточная монохроматизация может вызвать отклонение закона и тем в большей степени, чем меньше монохроматизация света.
2. В растворах могут протекать различные процессы, которые изменяют концентрацию поглощающего вещества или его природу: гидролиз, ионизация, гидратация, ассоциация, полимеризация, комплексообразование и др.
3. Светопоглощение растворов существенно зависит от рН раствора. При изменении рН раствора могут изменяться:
Степень ионизации слабого электролита;
Форма существования ионов, что приводит к изменению светопоглощения;
Состав образующихся окрашенных комплексных соединений.
Поэтому закон справедлив для сильно разбавленных растворов, и область его применения ограничена.
Визуальная колориметрия
Интенсивность окраски растворов можно измерять различными методами. Среди них выделяют субъективные (визуальные) методы колориметрии и объективные, то есть фотоколориметрические.
Визуальными называют такие методы, при которых оценку интенсивности окраски испытуемого раствора делают невооруженным глазом. При объективных методах колориметрического определения для измерения интенсивности окраски испытуемого раствора вместо непосредственного наблюдения пользуются фотоэлементами. Определение в этом случае проводят в специальных приборах - фотоколориметрах, поэтому метод получил название фотоколориметрического.
Цвета видимого излучения:
К визуальным методам относятся:
- метод стандартных серий;
- метод колориметрического титрования, или дублирования;
- метод уравнивания.
Метод стандартных серий. При выполнении анализа методом стандартных серий интенсивность окраски анализируемого окрашенного раствора сравнивают с окрасками серии специально приготовленных стандартных растворов (при одинаковой толщине слоя).
Метод колориметрического титрования (дублирования) основан на сравнении окраски анализируемого раствора с окраской другого раствора - контрольного. Контрольный раствор содержит все компоненты исследуемого раствора, за исключением определяемого вещества, и все использовавшиеся при подготовке пробы реактивы. К нему добавляют из бюретки стандартный раствор определяемого вещества. Когда этого раствора будет добавлено столько, что интенсивности окраски контрольного и анализируемого растворов уравняются, считают, что в анализируемом растворе содержится столько же определяемого вещества, сколько его было введено в контрольный раствор.
Метод уравнивания отличается от описанных выше визуальных колориметрических методов, в которых подобие окрасок стандартного и испытуемого растворов достигается изменением их концентрации. В методе уравнивания подобие окрасок достигается изменением толщины слоев окрашенных растворов. Для этой цели при определении концентрации веществ используют колориметры сливания и погружения.
Достоинства визуальных методов колориметрического анализа:
Техника определения проста, нет необходимости в сложном дорогостоящем оборудовании;
Глаз наблюдателя может оценивать не только интенсивность, но и оттенки окраски растворов.
Недостатки:
Необходимо готовить стандартный раствор или серии стандартных растворов;
Невозможно сравнивать интенсивность окраски раствора в присутствии других окрашенных веществ;
При длительном сравнивании интенсивности окраски глаз человека утомляется, и ошибка определения увеличивается;
Глаз человека не столь чувствителен к небольшим изменениям оптической плотности, как фотоэлектрические устройства, вследствие этого невозможно обнаружить разницу в концентрации примерно до пяти относительных процентов.
Фотоэлектроколориметрические методы
Фотоэлектроколориметрия применяется для измерения поглощения света или пропускания окрашенными растворами. Приборы, используемые для этой цели, называются фотоэлектроколориметрами (ФЭК).
Фотоэлектрические методы измерения интенсивности окраски связаны с использованием фотоэлементов. В отличие от приборов, в которых сравнение окрасок производится визуально, в фотоэлектроколориметрах приемником световой энергии является прибор - фотоэлемент. В этом приборе световая энергия преобразует в электрическую. Фотоэлементы позволяют проводить колориметрические определения не только в видимой, но также в УФ- и ИК-областях спектра. Измерение световых потоков с помощью фотоэлектрических фотометров более точно и не зависит от особенностей глаза наблюдателя. Применение фотоэлементов позволяет автоматизировать определение концентрации веществ в химическом контроле технологических процессов. Вследствие этого фотоэлектрическая колориметрия значительно шире используется в практике заводских лабораторий, чем визуальная.
На рис. 1 показан обычный порядок расположения узлов в приборах для измерения пропускания или поглощения растворов.
Рис.1 Основные узлы приборов для измерения поглощения излучения: 1 - источник излучения; 2 - монохроматор; 3 - кюветы для растворов; 4 - преобразователь; 5 - индикатор сигнала.
Фотоколориметры в зависимости от числа используемых при измерениях фотоэлементов делятся на две группы: однолучевые (одноплечие) - приборы с одним фотоэлементом и двухлучевые (двуплечие) - с двумя фотоэлементами.
Точность измерений, получаемая на однолучевых ФЭК, невелика. В заводских и научных лабораториях наиболее широкое распространение получил фотоэлектрические установки, снабженные двумя фотоэлементами. В основу конструкции этих приборов положен принцип уравнивания интенсивности двух световых пучков при помощи переменной щелевой диафрагмы, то есть принцип оптической компенсации двух световых потоков путем изменений раскрытия зрачка диафрагмы.
Принципиальная схема прибора представлена на рис. 2. Свет от лампы накаливания 1 с помощью зеркал 2 разделяется на два параллельных пучка. Эти световые пучки проходят через светофильтры 3, кюветы с растворами 4 и попадают на фотоэлементы 6 и 6", которые включены на гальванометр 8 по дифференциaльнoй схеме. Щелевая диафрагма 5 изменяет интенсивность светового потока, падающего на фотоэлемент 6. Фотометрический нейтральный клин 7 служит для ослабления светового потока, падающего на фотоэлемент 6".
Рис.2. Схема двухлучевого фотоэлектроколориметра
Определение концентрации в фотоэлектроколориметрии
Для определения концентрации анализируемых веществ в фотоэлектроколориметрии применяют:
Метод сравнения оптических плотностей стандартного и исследуемого окрашенных растворов;
Метод определения по среднему значению молярного коэффициента светопоглощения;
Метод градуировочного графика;
Метод добавок.
Метод сравнения оптических плотностей стандартного и исследуемого окрашенных растворов
Для определения готовят эталонный раствор определяемогo вещества известной концентрации, которая приближается к концентрацииисследуемого раствора. Определяют оптическую плотность этого раствора при определенной длине волны D эт. Затем определяют оптическую плотность исследуемого раствора D х при той же длине волны и при той же толщине слоя. Сравнивая значения оптических плотностей исследуемого и эталонного растворов, находят неизвестную концентрацию определяемого вещества.
Метод сравнения применим при однократных анализах и требует обязательного соблюдения основного закона светопоглощения.
Метод градуировочноro графика. Для определения концентрации вещества этим методом готовят серию из 5-8 стандартных растворов различной концентрации. При выборе интервала концентраций стандартных растворов руководствуются следующими положениями:
* он должен охватывать область возможных измерений концентрации исследуемого раствора;
* оптическая плотность исследуемого раствора должна соответствовать примерно середине градуировочной кривой;
* желательно, чтобы в этом интервале концентраций соблюдался основной закон светопоглощения, то есть график зависимости был прямолинейным;
* величина оптической плотности должна находиться в пределах 0,14… 1,3.
Измеряют оптическую плотность стандартных растворов и строят график зависимости D(С) . Определив D х исследуемого раствора, по градуировочному графику находят С х (рис. 3).
Этот метод позволяет определить концентрацию вещества даже в тех случаях, когда основной закон светопоглощения не соблюдается. В таком случае готовят большое количество стандартных растворов, отличающихся по концентрации не более чем на 10 %.
Рис. 3. Зависимость оптической плотности раствора от концентрации (калибровочная кривая)
Метод добавок - это разновидность метода сравнения, осно-ванный на сравнении оптической плотности исследуемого раствора и того же раствора с добавкой известно количества определяемого вещества.
Применяют его для устранения мешающего влияния посторонних примесей, определения малых количеств анализируемого вещества в присутствии больших количеств посторонних веществ. Метод требует обязательного соблюдения основного закона свето-поглощения.
Спектрофотометрия
Это метод фотометрического анализа, в котором определение содержания вещества производят по поглощению им монохроматического света в видимой, УФ- и ИК-областях спектра. В спектрофотометрии, в отличие от фотометрии, монохроматизация обеспечивается не светофильтрами, а монохроматорами, позволяющими непрерывно изменять длину волны. В качестве монохроматоров используют призмы или дифракционные решетки, которые обеспечивают значительно более высокую монохроматичность света, чем светофильтры, поэтому точность спектрофотометрических определений выше.
Спектрофотометрические методы, по сравнению с фотоколориметрическими, позволяют решать более широкий круг задач:
* проводить количественное определение веществ в широком интервал длин волн (185-1100 нм);
* осуществлять количественный анализ многокомпонентных систем (одновременное определение нескольких веществ);
* определять состав и константы устойчивости светопоглощающих комплексных соединений;
* определять фотометрические характеристики светопоглощающих соединений.
В отличие от фотометров монохроматором в спектрофо-тометрах служит призма или дифракционная решетка, позволяя-ющая непрерывно менять длину волны. Существуют приборы для измерений в видимой, УФ- и ИК-областях спектра. Принципи-альная схема спектрофотометра практически не зависит от спектральной области.
Спектрофотометры, как и фотометры, бывают одно- и двулучевые. В двулучевых приборах световой поток каким-либо способом раздваивают или внутри монохроматора, или по выходе из него: один поток затем проходит через испытуемый раствор, другой - через растворитель.
Однолучевые приборы особенно удобны при выполнении количественных определений, основанных на измерении оптической плотности при одной длине волны. В этом случае простота прибора и легкость эксплуатации представляют существенное преимущество. Большая скорость и удобство измерения при работе с двулучевыми приборами полезны в качественном анализе, когда для получения спектра оптическая плотность должна быть измерена в большом интервале длин волн. Кроме того, двулучевое устройство легко приспособить для автоматической записи непрерывно меняющейся оптической плотности: во всех современных регистрирующих спектрофото-метрах для этой цели используют именно двулучевую систему.
И одно-, и двулучевые приборы пригодны для измерений видимого и УФ-излучений. В основе ИК-спектрофотометров, выпускаемых промышленностью, всегда лежит двулучевая схема, поскольку их обычно используют для развертки и записи большой области спектра.
Количественный анализ однокомпонентных систем проводится теми же методами, что и в фотоэлектроколориметрии:
Методом сравнения оптических плотностей стандартного и исследуемого растворов;
Методом определения по среднему значению молярного коэффициента светопоглощения;
Методом градуировочного графика,
и не имеет никаких отличительных особенностей.
Спектрофотометрия в качественном анализе
Качественный анализ в ультрафиолетовой части спектра. Ультрафиолетовые спектры поглощения обычно имеют две-три, иногда пять и более полос поглощения. Для однозначной идентификации исследуемого вещества записывают его спектр поглощения в различных растворителях и сравнивают полученные данные с соответствующими спектрами сходных веществ известного состава. Если спектры поглощения исследуемого вещества в разных paстворителях совпадают со спектром известного вещества, то можно с большой долей вероятности сделать заключение об идентичности химического состава этих соединений. Для идентификации неизвестного вещества по его спектру поглощения необходимо располагать достаточным количеством спектров поглощения органических и неорганических веществ. Существуют атласы, в которых приведены спектры поглощения очень многих, в основном органических веществ. Особенно хорошо изучены ультрафиолетовые спектры аромати-ческих углеводородов.
При идентификации неизвестных соединений следует также обратить внимание на интенсивность поглощения. Очень многие органические соединения обладают полосами поглощения, максимумы которых расположены при одинаковой длине волны л, но интенсивность их различна. Например, в спектре фенола наблюдается полоса поглощения при л = 255 нм, для которой молярный коэффициент поглощения при максимуме поглощения е mах = 1450. При той же длине волны ацетон имеет полосу, для которой е mах = 17.
Качественный анализ в видимой части спектра. Идентификацию окрашенного вещества, например красителя, также можно проводить, сравнивая его спектр поглощения в видимой части со спектром сходного красителя. Спектры поглощения большинства красителей описаны в специальных атласах и руководствах. По спектру поглощения красителя можно сделать заключение о чистоте красителя, потому что в спектре примесей имеется ряд полос поглощения, которые отсутствуют в спектре красителя. По спектру поглощения смеси красителей можно также сделать заключение о составе смеси, особенно если в спектрах компонентов смеси имеются полосы поглощения, расположенные в разных областях спектра.
Качественный анализ в инфракрасной области спектра
Поглощение ИК-излучения связано с увеличением колебательной и вращательной энергий ковалентной связи, если оно приводит к изменению дипольного момента молекулы. Это значит, что почти все молекулы с ковалентными связями в той или иной мере способны к поглощению в ИК-области.
Инфракрасные спектры многоатомных ковалентных соединений обычно очень сложны: они состоят из множества узких полос поглощения и сильно отличаются от обычных УФ- и видимых спектров. Различия вытекают из природы взаимодействия поглощающих молекул и их окружения. Это взаимодействие (в конденсированных фазах) влияет на электронные переходы в хромофоре, поэтому линии поглощения уширяются и стремятся слиться в широкие полосы поглощения. В ИК -спектре, наоборот, частота и коэффициент поглощения, соответствующие отдельной связи, обычно мало меняются с изменением окружения (в том числе с изменением остальных частей молекулы). Линии тоже расширяются, но не настолько, чтобы слиться в полосу.
Обычно по оси ординат при построении ИК-спектров откладывают пропускание в процентах, а не оптическую плотность. При таком способе построения полосы поглощения выглядят как впадины на кривой, а не как максимумы на УФ-спектрах.
Образование инфракрасных спектров связано с энергией колебаний молекул. Колебания могут быть направлены вдоль валентной связи между атомами молекулы, в таком случае они называются валентными. Различают симметричные валентные колебания, в которых атомы колеблются в одинаковых направлениях, и асиммeтpичныe валентные колебания, в которых атомы колеблются в противоположных направлениях. Если колебания атомов происходят с изменением угла между связями, они называются деформационными. Такое разделение весьма условно, потому что при валентных колебаниях происходит в той или иной степени деформация углов и наоборот. Энергия деформационных колебаний обычно меньше, чем энергия валентных колебаний, и полосы поглощения, обусловленные деформационными колебаниями, располагаются в области более длинных волн.
Колебания всех атомов молекулы обусловливают полосы поглощения, индивидуальные для молекул данного вещества. Но среди этих колебаний можно выделить колебания групп атомов, которые слабо связаны с колебаниями атомов остальной части молекулы. Полосы поглощения, обусловленные такими колебаниями, называют характеристическими полосами. Они наблюдаются, как правило, в спектрах всех молекул, в которых имеются данные группы атомов. Примером характеристических полос могут служить полосы 2960 и 2870 см -1 . Первая полоса обусловлена асимметричными валентными колебаниями связи С-Н в метильной группе СН 3 , а вторая - симметричными валентными колебаниями связи С-Н этой же группы. Такие полосы с небольшим отклонением (±10 см -1) наблюдаются в спектрах всех насыщенных углеводородов и вообще в спектре всех молекул, в которых имеются СН 3 - группы.
Другие функциональные группы могут влиять на положение характеристической полосы, причем разность частот может составлять до ±100 см -1 , но такие случаи немногочисленны, и их можно учитывать на основании литературных данных.
Качественный анализ в инфракрасной области спектра проводится двумя способами.
1. Снимают спектр неизвестного вещества в области 5000-500 см -1 (2 - 20 мк) и отыскивают сходный спектр в специальных каталогах или таблицах. (или при помощи компьютерных баз данных)
2. В спектре исследуемого вещества отыскивают характеристические полосы, по которым можно судить о составе вещества.
Подобные документы
Изучение физико-химических методов анализа. Методы основанные на использовании магнитного поля. Теория методов по спектрометрии и фотоколореметрии в видимой области спектра. Спектрометрические и фотоколореметрические методы анализа лекарственных средств.
курсовая работа , добавлен 17.08.2010
Рефрактометрия как один из методов идентификации химических соединений, их количественного и структурного анализа, определения физико-химических параметров. Актуальность рефрактометрии для анализа лекарственных веществ для среднестатистической аптеки.
курсовая работа , добавлен 02.06.2011
Общее понятие о стероидах - производных ряда углеводородов, главным образом прегнана, андростана, эстрана. Лекарственные формы стероидных препаратов, их физико-химические свойства. Начало применения глюкокортикоидов в качестве лекарственных средств.
дипломная работа , добавлен 02.02.2016
Изучение номенклатуры лекарственных средств как источника информации для провизора. Информация о физико-химических свойствах препаратов. Длительность терапевтического эффекта. Лингвистический анализ номенклатуры ЛС. Закон о лекарственных средствах.
курсовая работа , добавлен 12.02.2015
Классификация лекарственных форм и особенности их анализа. Количественные методы анализа однокомпонентных и многокомпонентных лекарственных форм. Физико-химические методы анализа без разделения компонентов смеси и после предварительного их разделения.
реферат , добавлен 16.11.2010
Взаимодействие химических соединений с электромагнитным излучением. Фотометрический метод анализа, обоснование эффективности его использования. Исследование возможности применения фотометрического анализа в контроле качества лекарственных средств.
курсовая работа , добавлен 26.05.2015
Специфические особенности фармацевтического анализа. Испытание на подлинность лекарственных препаратов. Источники и причины недоброкачественности лекарственных веществ. Классификация и характеристика методов контроля качества лекарственных веществ.
реферат , добавлен 19.09.2010
Внутриаптечный контроль качества лекарственных средств. Химические и физико-химические методы анализа, количественное определение, стандартизация, оценка качества. Расчет относительной и абсолютной ошибок в титриметрическом анализе лекарственных форм.
курсовая работа , добавлен 12.01.2016
Применение антибиотиков в медицине. Оценка качества, хранение и отпуск лекарственных форм. Химические строение и физико-химические свойства пенициллина, тетрациклина и стрептомицина. Основы фармацевтического анализа. Методы количественного определения.
курсовая работа , добавлен 24.05.2014
Физико-химические процессы, возникающие при неправильном хранении лекарственных средств. Специфика химических, биологических процессов при воздействии различных факторов. Зависимость стабильности лекарственных веществ от условий хранения и получения.
Фармацевтический анализ (ФА). Он является основой фармацевтической химии и имеет свои особенности, отличающие его от других видов анализа. Они заключаются в том, что анализу подвергаются вещества различной химической природы: неорганические, элементоорганические, радиоактивные, органические соединения от простых алифатических до сложных природных БАВ. Чрезвычайно широк диапазон концентраций анализируемых веществ. Объектами фармацевтического анализа являются не только индивидуальные лекарственные вещества, но и смеси, содержащие различное число компонентов.
Ежегодное пополнение арсенала лекарственных средств вызывает необходимость разработки новых способов их анализа. Способы фармацевтического анализа нуждаются в систематическом совершенствовании в связи с непрерывным повышением требований как к качеству лекарственных средств, так и к количественному содержанию в них БАВ. Вот почему к фармацевтическому анализу предъявляют высокие требования. Он должен быть достаточно специфичен и чувствителен, точен по отношению к нормативным требованиям Государственной фармакопеи X и XI и другой НТД (ФС, ГОСТ), выполняться в короткие промежутки времени с использованием минимальных количеств испытуемых препаратов и реактивов.
В зависимости от поставленных задач фармацевтический анализ включает различные формы контроля качества лекарственных средств: фармакопейный анализ; постадийный контроль производства лекарств; анализ лекарственных форм индивидуального изготовления; экспресс-анализ в условиях аптеки и биофармацевтический анализ. Составной его частью является фармакопейный анализ, который представляет собой совокупность способов исследований лекарственных препаратов и лекарственных форм, изложенных в Государственной фармакопее или другой НТД (ФС, ФСП, ГОСТ). На основании результатов, полученных при выполнении фармакопейного анализа, делается заключение о соответствии лекарственного средства требованиям Государственной фармакопеи или другой НТД. При отклонении от этих требований лекарство не допускается к применению.
Химический анализ растительного сырья. По технике выполнения и характеру получаемых результатов химические реакции делят на несколько групп: качественные, микрохимические и гистохимические, микросублимация.
Для установления подлинности лекарственного растительного сырья используют простейшие качественные реакции и хроматографические пробы на действующие и сопутствующие вещества. Методика изложена в соответствующей нормативной документации на исследуемый вид сырья в разделе «Качественные реакции».
Качественные реакции выполняют на сухом сырье с такими видами сырья: кора дуба, калины, крушины, корневища бадана, корневища и корни девясила, корни одуванчика, алтея, женьшеня, барбариса, цветки липы, семена льна, склероции спорыньи (всего для 12 видов сырья).
В основном качественные реакции проводят с извлечением (вытяжкой) из лекарственного растительного сырья.
Исходя из свойств биологически активных веществ, их извлекают из сырья водой, спиртом различной концентрации или органическим растворителем, реже с добавлением щелочи или кислоты.
Водное извлечение готовят из сырья, содержащего гликозиды, полисахариды, сапонины, фенологликозиды, антрагликозиды, дубильные вещества. Подкисленной водой извлекают из сырья алкалоиды в виде солей.
Большую группу биологически активных веществ (сердечные гликозиды, кумарины, лигнаны, флавоноиды) извлекают этиловым и метиловым спиртом различной концентрации.
Если реакция достаточно специфична и чувствительна, то ее проводят с неочищенным экстрактом из сырья.
К таким реакциям относятся:
общеалкалоидные осадочные реакции;
реакции с раствором хлорида алюминия на флавоноиды (трава зверобоя, горца птичьего, горца перечного и др.);
проба Синода на флавоноиды в цветках бессмертника;
реакция с раствором щелочи на антраценпроизводные (кора крушины, корни ревеня и др.);
реакция с раствором железоаммонийных квасцов на дубильные веществ (кора дуба, корневища змеевика, бадана и др.).
Часто проведению реакции мешают сопутствующие вещества (белки, амины, стерины, хлорофилл). В этом случае используют очищенное извлечение (например, из сырья, содержащего сердечные гликозиды, кумарины, алкалоиды, фенологликозиды, лигнаны).
Очищают извлечение осаждением сопутствующих веществ раствором ацетата свинца и сульфата натрия или используют прием смены растворителей либо метод распределительной хроматографии.
Микрохимические реакции проводят обычно одновременно с микроскопическим анализом, наблюдая результаты под микроскопом:
на эфирное и жирное масло с раствором Судан III;
на одревесневшие лигнифицированные элементы с раствором флороглюцина и 25%-ным раствором серной кислоты или концентрированной хлороводородной кислоты.
На кору дуба (порошок) проводят реакцию с железоаммонийными квасцами и результат реакции изучают под микроскопом.
Гистохимические реакции - это такие реакции, с помощью которых можно выявить те или иные соединения непосредственно в клетках или структурах, где они локализуются.
По Государственной фармакопее XI, гистохимические реакции проводят на слизь с раствором туши в корнях алтея и семенах льна.
Микросублимация - непосредственное выделение из сухого растительного материала веществ, которые легко возгоняются при нагревании. Полученный сублимат исследуют под микроскопом, затем проводят микрохимическую реакцию с соответствующим реактивом.
Методы определения подлинности лекарственного растительного сырья. Подлинность сырья определяется макроскопическим, микроскопическим, химическим и люминесцентным анализами.
Макроскопический анализ. Для его проведения следует знать морфологию растений. Изучают внешний вид сырья невооруженным глазом или с помощью лупы, измеряют размеры частиц с помощью миллиметровой линейки. При дневном освещении определяют цвет сырья с поверхности, на изломе и на разрезе. Запах устанавливают при растирании или разломе растений, а вкус - только у неядовитых растений. При изучении внешнего вида обращают внимание на морфологические признаки частей сырья.
Микроскопический анализ. Используют для определения подлинности измельченного лекарственного растительного сырья. Для этого нужно знать анатомическую структуру растений в целом и характерные для конкретного растения признаки, отличающие его от других растений.
Химический анализ. Предусматривает проведение качественных, микрохимических, гистохимических реакций и сублимации для определения в сырье действующих или сопутствующих веществ. Микрохимические реакции целесообразно проводить параллельно с микроскопическим анализом. Гистохимические реакции проводят для выявления конкретных соединений в местах их локализации в растении. Под сублимацией понимают получение из растительного сырья легко возгоняемых при нагревании веществ с последующей качественной реакцией с сублиматом.
Люминесцентный анализ. Это метод исследования различных объектов (в том числе и биологических), основанный на наблюдении их люминесценции. Люминесценция - свечение газа, жидкости или твердого тела, обусловленное не нагревом тела, а нетепловым возбуждением его атомов и молекул. Люминесцентный анализ проводят для определения в лекарственном сырье веществ, обладающих люминесценцией.
Контроль качества органотерапевтических препаратов. Для проверки соответствия качества желез требованиям стандарта от каждой партии отбирают 5 % ящиков или пакетов, но не менее пяти таких упаковок. Если в одном из вскрытых ящиков или пакетов железы не соответствуют требованиям соответствующего стандарта хотя бы по одному из показателей, то проверяют всю партию.
Для единичных видов сырья имеются объективные (лабораторные) методы оценки его качества.
Объективно качество поджелудочной железы, предназначенной для производства инсулина, согласно ГОСТу, определяют по показателям массовой доли жира и массовой доли инсулина с помощью соответствующих лабораторных методов.
Массовую долю жира определяют жиромером. Массовую долю инсулина проверяют по требованию потребителя иммунореактивным методом с помощью антисыворотки, иммуноглобулинов в гомогенизированной железе.
Качество слизистой оболочки (эпителия) языков крупного рогатого скота проверяют путем определения величины pH консервирующей среды с эпителием и ее бактериальной обсемененности. Сущность метода заключается в определении общего количества микробов в 1 мл консервирующей среды с эпителием.
Качество стекловидного тела глаз крупного рогатого скота, свиней, овец и коз замороженного определяют по количественному содержанию гиалуроновой кислоты (по глюкозамину) в стекловидном теле. Принцип метода основан на определении глюкоза-мина в продуктах гидролиза гиалуроновой кислоты, который является составной частью молекулы гиалуроновой кислоты и находится в прямой зависимости от содержания его в стекловидном теле.
Биологическую активность гипофизов определяют в единицах действия АКТГ, содержащегося в 1 мг кислого ацетонированного порошка (КАП), полученного из гипофизов.
Определение активности АКТГ основано на его способности вызывать редукцию лимфоидной ткани, в частности зобной железы крысят. За единицу действия препарата принимают ту ежедневную дозу препарата, которая при введении в течение пяти суток вызывает уменьшение массы железы на 50±5 %.
Качество паращитовидных желез определяют гистологическим методом. На срезах паращитовидных желез просматриваются скопления эпителиальных клеток с выраженной базофильной зернистостью. На срезах лимфатических желез просматривается ретикулярная ткань (в виде однородной массы), окруженная плотной соединительной оболочкой (капсулой), от которой внутрь отходят ясно видимые соединительные тяжи. Государственным стандартом предусмотрено, что в пробе из 40 желез может содержаться не более одного лимфатического узла.
Методы определения качества сухих биологических препаратов. Сухие биологические препараты имеют ряд преимуществ по сравнению с традиционными жидкими биопрепаратами благодаря лучшему качеству, меньшей массе, возросшему сроку хранения, удобству транспортирования.
Физические методы. 1.Метод определения вакуума. Сущность метода заключается в способности высокочастотного электрического тока при большом напряжении вызывать в газах свечение, характер которого изменяется в зависимости от степени разреженности воздуха в ампуле (флаконе).
Отбор проб. Отбор проб проводят в соответствии с правилами, установленными в государственных стандартах на сухие биологические препараты.
Аппаратура и оборудование. При проведении испытания используют: аппарат типа «Д’Арсеналь» или «Тесла», штатив для ампул, стол металлический.
Проведение испытания. Подготовка к испытанию:
перед испытанием проверяют внешний вид, плотность укупоривания флаконов, наличие трещин, запайку ампул.
Аппарат выдерживают в течение 10 мин после включения. Испытуемые ампулы устанавливают в штативе, затем к ним подводят электрод на расстояние 1 см. При определении вакуума с помощью аппарата «Тесла» один металлический электрод аппарата заземляют через металлический стол, на котором разложены ампулы, а другой подводят к проверяемым ампулам. Экспозиция не более 1 с.
Обработка результатов. Появление свечения внутри ампул с характерным потрескиванием указывает на наличие в них вакуума.
Степень разрежения воздуха в проверяемых ампулах определяют по характеру свечения газов в проверяемых ампулах в соответствии с нижеследующими данными.
Определение степени разрежения воздуха в проверяемых ампулах
2. Метод определения в л а ж н о с т и. Сущность метода заключается в определении уменьшения массы пробы препарата после ее высушивания в течение 1 ч при температуре 105 °С.
Отбор проб. Для испытания из разных мест упаковки отбирают необходимое количество ампул (флаконов) с учетом требований к массе проб (в соответствии со стандартом).
При отборе проб проверяют герметичность ампул. У флаконов с лиофилизированным препаратом проверяют стенку и дно на целостность, а также полноту прилегания закатанного колпачка и резиновой пробки. При наличии дефектов флакон заменяют другим. Каждую ампулу, запаянную под вакуумом, перед извлечением из нее препарата проверяют на герметичность.
Аппаратура, материалы и реактивы. При проведении испытания используют: весы лабораторные, шкаф сушильный лабораторный, термометры ртутные, эксикатор, бюксы стеклянные, вазелин технический, кальций хлористый безводный или гипс обезвоженный, или силикагель прокаленный.
Подготовка к испытанию. Сушильный шкаф проверяют максимальными термометрами на равномерность нагрева.
При высушивании проб в бюксах нижняя часть контрольного термометра должна находиться на уровне бюкс. Показания контрольного термометра являются определяющими для настройки температуры в шкафу.
Весы должны быть установлены на прочном столе без вибрации. Результаты всех взвешиваний регистрируют в граммах с точностью до четвертого десятичного знака.
Нижняя часть эксикатора должна быть заполнена обезвоженным хлористым кальцием или гипсом, или силикагелем. Пришлифованные края сосуда слегка смазывают техническим вазелином.
Для каждого анализа должны быть подготовлены три бюксы одинаковых диаметров и высоты.
Проведение испытания. Для определения влажности используют три ампулы, если в каждой из них масса пробы не менее 0,1 г. Если ампула содержит менее 0,1 г биологического препарата, то можно использовать две и более ампул.
Отобранную пробу, растолченную до порошкообразного состояния, помещают ровным слоем в предварительно взвешенную бюксу.
Бюксы устанавливают в сушильный шкаф на полку. Началом сушки следует считать время достижения температуры 105 °С по контрольному термометру. Продолжительность сушки 60 мин.
После окончания сушки бюксы быстро закрывают крышками и переносят в эксикатор для охлаждения до комнатной температуры, после чего бюксы взвешивают с точностью до четвертого знака и регистрируют по форме.
3. Метод определения количества кислорода. Отбор проб. Отбор проб проводят в соответствии с правилами, установленными в государственных стандартах на сухие биологические препараты.
Аппаратура, материалы и реактивы. При проведении испытания используют: хроматограф газовый марки ЛXM-8МД или других аналогичных марок с детектором по теплопроводимости и газохромографической колонкой диаметром 3 мм и длиной 1000 мм, печь муфельную с температурой нагрева до 1000 °С, измеритель расхода газа с бюреткой, секундомер, шприц медицинский вместимостью 1 см 3 , сетки проволочные тканые, лупу измерительную, эксикатор, ступку фарфоровую, линейку металлическую длиной 30 см, сита молекулярные - цеолит синтетический марки СаА, иглу медицинскую, трубку медицинскую резиновую внутренним диаметром 4,2 мм, длиной 10 м, бутыль вместимостью 3000 см 3 , пробку резиновую, масло силиконовое, гелий, азот газообразный, воду дистиллированную.
Подготовка к испытанию. Подготовка колонки. Синтетический цеолит измельчают в фарфоровой ступке, отсеивают на ситах, промывают дистиллированной водой, высушивают и прокаливают в муфельной печи при температуре 450...500 °С в течение 2 ч, затем охлаждают в эксикаторе на сетках до комнатной температуры.
Хроматографическую колонку устанавливают вертикально и засыпают синтетическим цеолитом. Колонку не досыпают на 1 см и закупоривают сеткой. Заполненную колонку устанавливают в термостате хроматографа и, не присоединяя к детектору, пропускают через нее поток гелия или азота в течение 3 ч при температуре 160... 180 °С. Затем колонку присоединяют к детектору и продолжают через нее пропускать гелий или азот, пока не прекратится дрейф нулевой линии при максимальной чувствительности детектора.
Подготовку хроматографа к работе и включение выполняют в соответствии с заводской инструкцией.
Подготовка флакона с препаратом к испытанию. Для отбора пробы из флакона с препаратом выравнивают давление газа во флаконе с атмосферным давлением.
Подготовка медицинского шприца. Предварительно устанавливают на штоке шприца металлическую трубку и проверяют шприц на герметичность. Проверенным и подготовленным к отбору газа медицинским шприцем с иглой прокалывают резиновую трубку, по которой выходит гелий из колонки сравнения хроматографа, и дважды медленно шприцем набирают и выпускают гелий. В третий раз, набрав гелий в шприц и расположив его иглой вниз, отбирают пробы газа из флакона с препаратом.
Проведение испытания. Из каждого флакона отбирают две пробы газа и последовательно одну за другой с интервалом 3...4 мин вводят в испаритель хроматографа. Пробу в испаритель вводят плавным нажатием пальца на шток. Через 110... 120 с после ввода пробы на хроматограмме самописец вычерчивает пик кислорода, а затем пик азота.
Обработка результатов. Рассчитывают площадь пиков кислорода и азота. Для этого на хроматографе измеряют высоту и ширину пиков кислорода и азота с помощью металлической линейки длиной 30 см, увеличительной лупы и остро заточенного карандаша. Высоту пиков измеряют от базовой линии до вершины пика, ширину пика - на половине его высоты. При измерениях берут расстояние от внутренней толщины линии пика до наружной.
Площадь пиков кислорода (SО 2 , мм 2) и азота (5N 2 , мм 2) вычисляют по формулам
SО 2 = h 1 *b 1 ; SN = h 2 *b 2 ,
где h 1 h 2 ~ высота пиков кислорода и азота, мм; b 1 , b 2 - ширина пиков кислорода и азота, мм.
Объемную долю кислорода (X, %) в каждой пробе газа вычисляют по формуле
X=SO 2 /(SO 2 +SN 2)
где SO 2 , SN 2 - площади пиков кислорода и азота, мм 2 .
За окончательный результат испытания принимают среднее арифметическое результатов определений в трех флаконах препарата.
Относительная приведенная погрешность метода при доверительной вероятности Р- 0,95 не должна превышать 10 %.
Бактериологический метод. Контроль стерильности. Сущность метода заключается в микробиологической оценке отсутствия роста бактерий и грибов в высевах препаратов на питательные среды.
Отбор проб. От каждой серии препаратов отбирают пробы в количестве 0,15 % флаконов, но не менее пяти для жидких и 10 ампул для сухих препаратов.
Подготовка к испытанию. Лабораторную посуду кипятят в течение 15 мин в дистиллированной воде, подкисленной раствором соляной кислоты, а затем промывают водопроводной водой и моют ершом в растворе, содержащем на 1000 см 3 дистиллированной воды 30 г стирального порошка и 50 см 3 водного аммиака. После этого посуду тщательно промывают сначала водопроводной водой, а затем три раза дистиллированной водой, высушивают и стерилизуют.
Перед стерилизацией посуду укладывают в металлические пеналы. Стерилизуют посуду в автоклаве при 0,15 МПа в течение 60 минут.
Готовые питательные среды, проверенные на ростовые свойства, разливают по 6...8 см 3 (для определения анаэробов по 10...12 см 3) в пробирки, по 50...60 см 3 во флаконы вместимостью 100 см 3 .
Пробы сухих биологических препаратов предварительно растворяют стерильным растворителем (изотонический раствор хлорида натрия, дистиллированная вода и т. д.).
Проведение испытания. 1. Проведение испытания на стерильность с использованием тиогликолевой среды.
Из каждого флакона препарата производят посев по 1 см 3 в три пробирки, содержащие тиогликолевую среду.
Две засеянные пробирки выдерживают в термостате в течение 14сут: одну -при температуре 21 °С, другую -при температуре 37 °С.
Третью пробирку выдерживают в течение 7 сут при температуре 37 °С и затем делают из нее пересевы по 0,5 см 3 по одной пробирке на скошенный казеиновый агар, казеиновый питательный бульон, среду Сабуро и по 1 см 3 на казеиновый питательный бульон под вазелиновым маслом с кусочками мяса или печени.
Пересевы на казеиновый агар, мясопептонный бульон выдерживают еще в течение 7 сут при температуре 37 °С, а пересев на среду Сабуро - при температуре 21 °С.
При испытании проб препаратов проводят контроль стерильности сред: три пробирки с каждой средой выдерживают в термостате в течение 14 сут при 37 °С, со средой Сабуро - при температуре 21 °С.
2. Проведение испытания на стерильность без тиогликолевой среды.
Из каждой пробы препарата производят посев на жидкую среду Сабуро, мясопептонный агар и мясопептонный бульон - по три пробирки; на среду Тароцци - по две пробирки и два флакона.
Для выявления аэробов высевают 0,5 см 3 посевного материала в одну пробирку и 1...2 см 3 в один флакон, а для выявления анаэробов - соответственно по 1 и 5 см 3 . Посевы помещают в термостат (при температуре 37 °С; для Сабуро - при температуре 21 °С) на 7 сут (15 сут для анаэробов). Затем делают пересев (кроме посевов на мясопептонном агаре). Пересевают на те же среды. Выдерживают 7 сут (15 сут для анаэробов). Проводят контроль стерильности.
Оценка результатов. Учитывают результаты первичного и повторного посевов путем макроскопического, а в случае роста микроорганизмов - микроскопического исследования всех посевов, учитывают через 14 сут после первичного посева на тиогликолевой среде и через 7 сут после первичного посева без тиогликолевой среды. Среду считают стерильной, если ни в одной из засеянных пробирок не наблюдается рост.
В случаях роста хотя бы в одной из засеянных пробирок контроль стерильности повторяют на том же количестве проб и проводят микроскопию выросших микробов. Мазки окрашивают по Граму, отмечая морфологию.
При отсутствии роста в повторном контроле препарат считают стерильным. При наличии роста хотя бы в одной из пробирок и идентичности микрофлоры при первичном и повторном посевах препарат считают нестерильным.
Если при первичном и повторном посевах выявлена различная микрофлора, а также выявлен рост лишь в отдельных пробирках, проводят посев образцов в третий раз.
При отсутствии роста препарат считают стерильным. При обнаружении роста хотя бы в одной пробирке независимо от характера микрофлоры препарат считают нестерильным.
Нормативные требования к качеству готовых лекарственных форм. Лекарственные формы изготовляют на заводах, фармацевтических фабриках (официальные лекарственные средства) и в аптеках (магистральные лекарственные средства). Контроль готовых лекарственных форм на фармацевтических предприятиях осуществляют в соответствии с требованиями НТД (Государственной фармакопеи, ФС, ФСП, ГОСТов). В соответствии с требованиями этих документов лекарственные формы должны подвергаться проверке (В. Д. Соколов, 2003).
Таблетки испытывают на распадаемость. Если нет других указаний в частной статье, то таблетки должны распадаться в течение 15 мин, а покрытые оболочкой не более 30 мин. Кишечнорастворимые таблетки не должны распадаться в течение 1 ч в растворе соляной кислоты, но должны распадаться в течение 1 ч в растворе натрия гидрокарбоната. Прочность таблеток на истирание должна быть не менее 75 %. Лекарственное средство, содержащееся в таблетке, должно растворяться в воде за 45 мин не менее чем на 75 %. Среднюю массу определяют взвешиванием 20 таблеток с точностью до 0,001 г. Допускаются отклонения от средней массы: ±7,5%-для таблеток массой 0,1...0,3 г и ±5%-для таблеток массой 0,5 г и более. В таблетках также контролируют содержание талька.
Гранулы - определяют размер с помощью ситового анализа. Диаметр ячейки должен быть 0,2...3 мм, а число более мелких и более крупных гранул не должно превышать 5 %. Испытание распадаемости гранул из навески 0,5 г такое же, как и у таблеток. Время распадаемости не должно превышать 15 мин. Определяют влагу. Для выявления содержания лекарственного вещества берут навеску не менее чем из 10 растертых гранул.
Капсулы - контролируют среднюю массу. Отклонение от нее каждой капсулы не должно превышать ±10 %. Подобно тому как это проводят с таблетками, контролируют распадаемость и растворимость, а также определяют однородность дозирования для капсул, содержащих 0,05 г и менее лекарственного вещества. Количественное определение лекарственных веществ выполняют по специальным методикам, используя для этих целей содержимое от 20 до 60 капсул.
Порошки - устанавливают отклонения в массе дозированных порошков. Они могут быть ±15% при массе порошка до 0,1 г; ±10 % - от 0,1 до 0,3 г; ±5 % - от 0,3 до 1; ±3 % - свыше 1 г.
Суппозитории - визуально определяют однородность на продольном разрезе. Среднюю массу устанавливают взвешиванием с точностью до 0,01 г, отклонения не должны превышать ± 5 %. Суппозитории, изготовленные на липофильных основаниях, контролируют по температуре плавления. Она не должна превышать
37 °С. Если эту температуру установить невозможно, то определяют время полной деформации, которое должно быть не более 15 мин. Суппозитории, изготовленные на гидрофильной основе, испытывают на растворимость (показатель «растворение»). Определяют время растворения при температуре (37±1) °С, которое не должно превышать 1 ч. Количественное определение лекарственных веществ проводят по специальным методикам.
Настойки - определяют содержание спирта или плотность. Содержание действующих веществ устанавливают с помощью специальных методик. Кроме того, определяют сухой остаток после выпаривания в бюксе 5 мл настойки досуха и высушивания его в течение 2 ч при температуре (102,5±2,5) °С. В таком же объеме настойки после сжигания и прокаливания ее смеси с 1 мл концентрированной серной кислоты определяют содержание тяжелых металлов.
Экстракты - как и в настойках, определяют плотность или содержание спирта, действующих веществ, тяжелых металлов. Устанавливают также сухую массу остатка, а в густых и сухих экстрактах - содержание влаги [высушиванием в сушильном шкафу при температуре (102,5±2,5) °С).
Аэрозоли - измеряют давление внутри баллона с помощью манометра при комнатной температуре (если пропеллентом служит сжатый газ). Проверяют упаковку на герметичность. В дозированных упаковках определяют среднюю массу препарата в одной дозе, отклонение в которой допускается не более +20 %. Устанавливают процент выхода содержимого путем удаления его из баллона с последующим взвешиванием. Количественное определение вещества проводят в соответствии с требованиями частных статей Государственной фармакопеи. Отклонения от изложенных количеств не должно превышать ±15 %.
Мази - общим испытанием является метод определения размера частиц лекарственного вещества в мазях. Используют микроскоп с окулярным микрометром МОВ-1.
Пластыри. Состав, показатели качества, методики испытаний бывают разные и изложены в нормативной документации на конкретную продукцию.
Капли глазные испытывают на стерильность и наличие механических включений.
Инъекционные лекарственные формы. Особого внимания требуют инъекционные лекарственные растворы, вводимые внутривенно в больших количествах. Используют такие характеристики, как внешний вид, в том числе окраска и прозрачность растворов, отсутствие механических примесей, апирогенность, стерильность, объем раствора, количество в нем действующего вещества, pH и изотоничность плазмы крови, упаковка, маркировка, объем наполнения ампул. Нормы допустимых отклонений указаны в Государственной фармакопее XI. Кроме того, определяют содержание вспомогательных веществ; для некоторых из них (фенол, крезол, сульфиты, хлорбутанол) предусмотрены допустимые количества (от 0,2 до 0,5 %). Требования к pH зависят от препарата, обычно его показатель может находиться в пределах от 3,0 до 8,0. На каждой ампуле (флаконе) указывают название лекарственного средства, его содержание (в процентах) или активность (в единицах действия, ЕД), объем или его массу, номер серии, срок годности. Проведение всех испытаний инъекционных лекарственных форм регламентировано НТД.
Анализ гомеопатических лекарственных средств весьма труден из-за высоких разведений лекарственных веществ. Если БАВ содержатся в настойках, эссенциях, мазях и других формах в разведениях до 2 С (С - сотенное) или 0,0001, то их анализ и стандартизация практически не отличаются от контроля качества лекарственных форм, используемых в аллопатической медицине. Лекарственные средства в разведении 2...3 С (10 -4 ...10 -6) анализируют после проведения специальных приемов концентрации с помощью упаривания, сжигания веществ с последующим определением одним из физико-химических методов, исходя из его разрешающей способности. При более чем 3 С разведении (10 -6) достаточно установить подлинность лекарственного средства, содержащегося в одной разовой или суточной дозе. При очень высоких разведениях (до 50 С или 10 -10 ...10 -100) контроль качества гомеопатических средств существующими методами выполнить невозможно. Для таких лекарств контроль качества осуществляют на стадии получения, строго контролируя технологический процесс. Качество контролируют при закладке ингредиентов и фиксируют в акте загрузки. Каждый ингредиент подвергают предварительному анализу. Во всех перечисленных случаях для анализа и стандартизации гомеопатических лекарственных средств используют хроматографические, фотометрические, флуоресцентные и другие методы.
 Могильники Чернобыля: радиоактивные отходы зоны отчуждения Где находится техника чернобыля
Могильники Чернобыля: радиоактивные отходы зоны отчуждения Где находится техника чернобыля Девочка заговорила на иностранном языке после комы
Девочка заговорила на иностранном языке после комы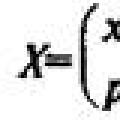 Сравнение законов распределения вероятностей
Сравнение законов распределения вероятностей