Скорость реакции нитрования ароматических соединений. Выбранный путь синтеза - анализ, описание методик выполнения стадий эксперимента
Поговорим о том, как осуществляется нитрование толуола. Получают подобным взаимодействием огромное количество полуфабрикатов, используемых в изготовлении взрывчатых веществ, фармацевтических препаратов.
Значимость нитрования
Производные бензола в виде ароматических нитросоединений выпускаются в современной химической промышленности. Нитробензол является полупродуктом в анилинокрасочном, парфюмерном, фармацевтическом производстве. Он является отличным растворителем для многих органических соединений, включая и нитрит целлюлозы, формируя с ним желатинообразную массу. В нефтяной промышленности его применяют в качестве очистителя для смазочных масел. При нитровании толуола получают бензидин, анилин, фенилендиамин.
Характеристика нитрования
Нитрование характеризуется вводом группы NO2 в молекулу органического соединения. В зависимости от исходного вещества протекает данный процесс по радикальному, нуклеофильному, электрофильному механизму. В качестве активных частиц выступают катионы нитрония, ионы и радикалы NO2. Реакция нитрования толуола относится к замещению. Для других органических веществ возможно заместительное нитрование, а также присоединение по двойной связи.
Нитрование толуола в молекуле ароматического углеводорода осуществляется с помощью нитрующей смеси (серной и азотной кислот). Каталитические свойства проявляет выступающая в данном процессе в качестве водотнимающего средства.

Уравнение процесса
Нитрование толуола предполагает замещение одного водородного атома нитрогруппой. Как выглядит схема протекающего процесса?
Для того чтобы описать нитрование толуола, уравнение реакции можно представить в следующем виде:
ArH + HONO2+ = Ar-NO2 +H2 O
Оно позволяет судить только об общем ходе взаимодействия, но не раскрывает всех особенностей данного процесса. На самом деле происходит реакция между ароматическими углеводородами и продуктами азотной кислоты.
Учитывая, что в продуктах есть молекулы воды, это приводит к снижению концентрации азотной кислоты, поэтому нитрование толуола замедляется. Для того чтобы избежать подобной проблемы, осуществляют данный процесс при невысоких температурах, используя азотную кислоту в избыточном количестве.
Помимо серной кислоты, в качестве водоотнимающих средств применяют полифосфорные кислоты, трехфтористый бор. Они дают возможность снизать расход азотной кислоты, повышают эффективность взаимодействия.

Нюансы процесса
Нитрование толуола было описано в конце девятнадцатого века В. Марковниковым. Ему удалось установить связь между присутствием в реакционной смеси и скоростью протекания процесса. В современном производстве нитротолуола применяют безводную азотную кислоту, взятую в некотором избытке.
Кроме того, сульфирование и нитрование толуола связано с использованием доступного водоотнимающего компонента фторида бора. Его введение в реакционный процесс позволяет снижать стоимость получаемого продукта, что делает доступным нитрование толуола. Уравнение протекающего процесса в общем виде представлено ниже:
ArH + HNO3 + BF3= Ar-NO2 + BF3 ·H2 O
После завершения взаимодействия вводят воду, благодаря чему моногидрат фторида бора образует дигидрат. Его отгоняют в вакууме, затем добавляют фтористый кальций, возвращая соединение в исходный вид.

Специфика нитрования
Есть некоторые особенности данного процесса, связанные с выбором реагентов, субстракта реакции. Рассмотрим некоторые их варианты подробнее:
- 60-65 процентная азотная кислота в смеси с 96 процентной серной кислотой;
- смесь 98 % азотной кислоты и концентрированной серной кислот подходит для мало реакционных органических веществ;
- нитрат калия или аммония с концентрированной серной кислотой - это отличный выбор для производства полимерных нитросоединений.

Кинетика нитрования
Взаимодействующие со смесью серной и азотной кислот, нитруются по ионному механизму. В. Марковникову удалось охарактеризовать специфику данного взаимодействия. Процесс протекает в несколько стадий. Сначала образуется нитросерная кислота, которая подвергается диссоциации в водном растворе. Ионы нитрония вступают во взаимодействие с толуолом, образуя в качестве продукта нитротолуол. При добавлении в смесь молекул воды происходит замедление процесса.
В растворителях с органической природой - нитрометане, ацетонитриле, сульфолане - образование этого катиона позволяет увеличивать скорость нитрования.
Полученный катион нитрония прикрепляется к ядру ароматического толуола, при этом образуется промежуточное соединение. Далее происходит отрыв протона, приводящий к формированию нитротолуола.
Для детального описания происходящего процесса можно рассмотреть образование «сигма» и «пи» комплексов. Образование «сигма» комплекса является лимитирующей стадией взаимодействия. будет напрямую связана с быстротой присоединения катиона нитрония к атому углерода в ядре ароматического соединения. Отщепление протона от толуола осуществляется практически мгновенно.
Только в некоторых ситуациях могут возникать какие-то проблемы с замещением, связанные с существенным первичным кинетическим изотопным эффектом. Это связано с ускорением обратного процесса при наличии препятствий разного вида.
При выборе в качестве катализатора и водоотнимающего средства концентрированной серной кислоты наблюдается смещение равновесия процесса в сторону образования продуктов реакции.

Заключение
При нитровании толуола образуется нитротолуол, который является является ценным продуктом химической промышленности. Именно это вещество является взрывчатым соединением, поэтому востребовано на взрывных работах. Среди экологических проблем, связанных с его промышленным изготовлением, отметим использование существенного количества концентрированной серной кислоты.
Для того чтобы справиться с такой проблемой, химики ищут способы уменьшения сернокислотных отходов, получаемых после проведения процесса нитрования. Например, процесс осуществляют при пониженных температурах, применяют легко регенерируемые среды. Серная кислота обладает сильными окислительными свойствами, что негативно отражается на коррозии металлов, представляет повышенную опасность для живых организмов. При соблюдении всех норм безопасности можно справиться с этими проблемами, получать нитросоединения высокого качества.
Нитрование
Реакция нитрования является одной из наиболее изученных реакций ароматического замещения. Для препаративных целей нитрование, как правило, проводят смесью концентрированных азотной и серной кислот, так называемой нитрующей смесью . На первой стадии реакции происходит образование иона нитрония + NO 2 , который и является электрофильным агентом:
HO-NO 2 + H 2 SO 4 H 2 O + -NO 2 + HSO 4 -
H 2 O + -NO 2 + H 2 SO 4 H 3 O + + HSO 4 - + + NO 2
Наличие иона нитрония в этом растворе подтверждено спектроскопически. Азотная кислота в концентрированной серной кислоте практически нацело превращается в нитроний-катион. Незначительная эффективность самой азотной кислоты в реакции нитрования бензола объясняется низким содержанием иона + NO 2 .
B качестве нитрующих агентов используются также другие системы, в которых генерируется либо катион + NO 2 , либо соединение общей формулы NO2-Y где Y - хорошая уходящая группа. Некоторые из таких систем, нашедшие наибольшее применение, представлены в таблице 1 в порядке увеличения их активности.
Таблица 1. Нитрующие реагенты.
| Нитрующий реагент | Метод генерации | Арены, подвергающиеся нитрованию |
| Азотная кислота HO-NO2 |
Фенолы, эфиры фенолов, бифенил | |
| Ацетилнитрат CH 3 C(O)-O-NO 2 | СН 3 СООН + HNO 3 (СН 3 СО) 2 О + HNO 3 | Бензол, алкилбензолы |
| Диоксид азота N 2 O 4 (O=N-O-NO 2) |
Бензол, алкилбензолы | |
| Нитрующая смесь | H 2 SO 4 конц + HNO 3 | Бензол, алкилбензолы, галогенбензолы, бензойная кислота, нитробензол, нафталин |
| Хлорид нитрония Сl-NO 2 |
Бензол, алкилбензолы, нитробензол, | |
| Тетрафторборат нитрония BF 4 -+ NO 2 | HF . 2BF 3 + HNO 3 | Динитробензол |
На примере реакции нитрования алкилбензолов отчетливо прослеживается влияние пространственных факторов на направление электрофильного замещения. Так, при нитровании толуола (метилбензола) орто -изомер образуется в качестве основного продукта, а при переходе к этил-, изо -пропил- и особенно к трет -бутил-бензолу его выход существенно уменьшается (см. табл. 2).
Таблица 2. Влияние пространственых факторов на соотношение орто-, пара-изомеров в реакции нитрования (NO 2 +)
Замещение в положения, % |
|||
C 6 H 5 -C 2 H 5 |
|||
C 6 H 5 -CH(CH 3) 2 |
|||
C 6 H 5 -C(CH 3) 3 |
|||
При изучении нитрования алкилбензолов было обнаружено так называемое ипсо-замещение , когда электрофильная атака протекает по тому атому углерода бензольного кольца, которое уже содержит заместитель, например:

В отличие от нитрования, при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены, например, Cl 2 и Br 2 ,(прим.35) могут легко атаковать активированное ароматическое ядро (например, фенола), но не способны реагировать с бензолами и алкилбензолами (фотохимическая активация может, однако, в последнем случае привести к протеканию радикального замещения в боковую цепь; см. раздел IV.3). Для поляризации атакующей молекулы галогена необходим катализ кислотами Льюиса , такими как AlCl 3 , FeBr 3 , и т.п.; при этом в молекуле галогена появляется так называемый "электрофильный конец" (энергия же, требующаяся для образования катиона Наl + существенно выше). Тем самым электрофильное замещение существенно облегчается:

Галогенирование протекает очень энергично, если использовать реагенты, в которых галоген в результате поляризации имеет сильный положительный заряд или даже существует как катион. Так, очень инертный мета -динитробензол можно пробромировать бромом в концентрированной серной кислоте в присутствии сульфата серебра. Предполагают, что в этом случае промежуточно образуется бром-катион:
2Br 2 + Ag 2 SO 4 2Br + + 2AgBr + SO 4 2-
Реакционная способность элементарного иода в реакциях электрофильного замещения в ароматическом ядре незначительна, так что прямое иодирование возможно только в случае фенола и ароматических аминов. Иодирование других ароматических соединений проводят в присутствии окислителя (обычно, азотной кислоты). Считается, что в этих условиях роль электрофильного агента играет ион I- + OH 2 .

Для галогенирования аренов можно применять также смешанные галогены , например, монохлорид брома (BrCl) или иода (ICl):

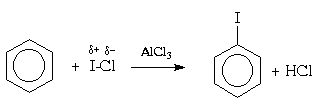
Галогенирование in vivo . В качестве примера электрофильного ароматического галогенирования, протекающего в живых организмах, можно привести реакцию иодирования -аминокислоты - тирозина в ходе биосинтеза иодсодержащих гормонов щитовидной железы до 3-иодтирозина и далее до 3,5-дииодтирозина:

Детали механизма сульфирования исследованы менее подробно по сравнению с нитрованием и галогенированием. Сам бензол сульфируется довольно медленно горячей концентрированной серной кислотой, но быстро - олеумом, SO 3 в инертных растворителях или комплексом SO 3 с пиридином. Природа электрофильной частицы зависит от условий реакции, но, вероятно, это всегда SO 3 , или в свободном состоянии, или связанный с "носителем", например, в виде H 2 SO 4 . SO 3 (H 2 S 2 O 7) в серной кислоте. Небольшие количества SO 3 образуются в H 2 SO 4:
2H 2 SO 4 SO 3 + H 3 O + + HSO 4 -
Атаку ароматического субстрата осуществляет атом серы поскольку он сильно положительно поляризован, то есть электронодефицитен:

Сульфирование является обратимым процессом. Это имеет практическое значение: при обработке сульфокислот водяным паромпроисходит замещение группы SO 3 Н на водород. Таким образом, можно ввести группу SO 3 Н как заместитель, ориентирующий требуемым образом последующие реакции (см. раздел IV.1.Б), а затем ее отщепить. Некоторые интересные особенности имеет сульфирование нафталина (см. раздел IV.1.Г).
Подобно галогенам, алкилгалогениды могут быть так сильно поляризованы кислотами Льюиса (хлоридами алюминия и цинка, трифторидом бора и др.), что они становятся способными к электрофильному замещению в ароматическом ядре:
R-Cl + AlCl 3 R +... Cl ...- AlCl 3 R + AlCl 4 -
Кроме алкилгалогенидов, источниками карбокатионов для галогенирования ароматических соединений могут быть алкены или спирты. При этом необходимо присутствие протонной кислоты, чтобы протонировать алкен или спирт. В случае спиртов требуется добавка не менее чем эквимольного количества кислоты (так как вода, выделяющаяся в ходе реакции, дезактивирует эквимольное количество катализатора), тогда как в реакциях с участием алкилгалогенидов и алкенов достаточно добавлять незначительное количество катализатора.
В лаборатории алкилирование по Фриделю-Крафтсу имеет ограниченное применение, так как обычно при этой реакции образуются смеси продуктов, что обусловлено рядом причин:
1) Образующийся продукт алкилирования легче вступает в реакции электрофильного ароматического замещения, чем исходное соединение (Alk - электронодонорная группа), поэтому дальше преимущественно алкилируется продукт. Если хотят получить продукты моноалкилирования, то необходимо брать большой избыток ароматического соединения.
2) Как и сульфирование, реакция алкилирования по Фриделю-Крафтсу обратима (см. также раздел IV.1.Г).
3) Даже в мягких условиях первичные и вторичные алкилгалогениды дают преимущественно вторичные или третичные алкиларены соответственно, поскольку алкилирование происходит в условиях, приближающихся к S N 1 реакции.(прим.37) Перегруппировки можно избежать, если работать при низких температурах.
Ацилирование ароматических соединений по Фриделю-Крафтсу является важнейшим методом синтеза жирноароматических кетонов. Производные карбоновых кислот, такие как ацилгалогениды и ангидриды, имеют полярную карбонильную группу и в принципе способны к электрофильному замещению в ароматических системах:

Электрофильная активность этих соединений, однако, невелика, и должна быть повышена действием кислот Льюиса. При этом кислотный катализатор, как правило, атакует атом кислорода карбонильного соединения и, смещая электронную плотность, повышает положительный заряд соседнего атома углерода. В результате образуется поляризованный комплекс (а в пределе - ацилкатион), действующий как электрофил:

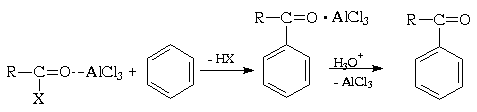
Важное отличие реакции ацилирования ацилгалогенидами от реакции алкилирования алкилгалогенидами состоит в том, что в первой из этих реакций требуется более 1 моль кислоты Льюиса , тогда как во второй необходимо только каталитические количество. Это обусловлено тем, что кислота Льюиса образует комплекс как с ацилирующим производным карбоновой кислоты, так и с кетоном - продуктом реакции. При взаимодействии с ангидридами получающаяся кислота связывает еще моль моль катализатора, так что в целом его необходимо по крайней мере два моль. В каждом случае по окончании реакции образовавшийся комплекс кетона с хлоридом алюминия (или другой кислотой Льюиса) должен быть гидролитически разрушен (соляной кислотой со льдом).
Полиацилирования не наблюдается, поскольку образующийся кетон значительно менее реакционноспособен, чем исходное соединение (см. раздел IV.1.Б). Поэтому алкилбензолы часто предпочитают получать не прямым алкилированием, а ацилированием по Фриделю-Крафтсу с последующим восстановлением. Ароматические соединения с сильнодезактивирующими заместителями, например, нитро- или циано- группами, также не ацилируются по Фриделю-Крафтсу.
Контрольные задачи
2. Изобразите диаграмму потенциальной энергии
для реакции электрофильного ароматического
замещения, в которой медленной стадией является
образование
-комплекса (например,
нитрование бензола борфторидом нитрония;
см. раздел IV.1.A).
3. Какой продукт преимущственно образуется при бромировании: а)пара -нитротолуола; б) мета -нитробензолсульфокислоты; в) орто -нитрофенола.
4. Адреналин (1-(3",4"-дигидроксифенил)-2-метиламиноэтанол)- первый гормон, выделенный из мозгового вещества надпочечников, в настоящее время синтезируюют в три стадии из пирокатехина. Напишите уравнение первой стадии этого синтеза - реакции ацилирования пирокатехина (1,2-дигидроксибензола) хлорангидридом хлоруксусной киcлоты и объясните механизм).
5. Одной их качественных реакций на белки является ксантопротеиновая реакция, указывающая на присутствие ароматических -аминокислот. Она заключается в обработке белка азотной кислотой при нагревании. Напишите уравнение ксантопротеиновой реакции с тирозином (см. раздел I), образовавшимся в результате гидролиза белка.
Получение через диазосоединение
Методы этой группы гораздо менее многочисленны, но отличаются высокими выходами, низкими содержаниями примесей побочных продуктов, простотой и разнообразием в исполнении.
Наиболее простым и надежным способом этой группы является проведение реакции Зандмейера. Можно привести 2 примера различного проведения только этой стадии:

2.3 Другие методы

PhBr + TfOMe, антраниловая кислота в реакции Бородина- Хунсдикера, реакция о-дибромбензола и MeMgBr и тд. - имеют преимуществ по сравнению с другими методами и отличаются более низкой препаративной ценностью, хотя и представляют интерес.
Выбранный путь синтеза - анализ, описание методик выполнения стадий эксперимента
Основным критерием выбора того или иного метода, подробно описанного выше, является надежность и доступность. Этому отвечает путь толуол - нитротолуол - о-толуидин - о-бромтолуол.
Нитрование толуола
В трехгорлую колбу на 250 мл, снабженную мешалкой, капельной воронкой, внутренним термометром (прибор не должен быть герметичен) помещают 0,15 моль ароматического нитросоединения. Затем медленно, при хорошем перемешивании и охлаждении баней со льдом добавляют нитрующую смесь, предварительно охлажденную до по меньшей мере до 10 °C, температура реакционной смеси должна находится в интервале 5-10 °C.
Далее при комнотной температуре перемешивают ещё 2-3 часа. После этого реакционную смесь осторожно выливают в 300 мл ледяной воды и хорошо перемешивают. Отделяют органический слой, водный экстрагируют эфиром. Объединённые органические вытяжки промывают водой, 2 н. раствором бикарбоната натрия до нейтральной реакции, потом опять водой. обогревателем. Вытяжки высушивают над CaCl 2 и перегоняют. П-изомер вымораживают смесью льда и соли, промывают небольшим количеством холодного петролейного эфира. (Тщательное разделение довольно, этот метод оставляет около 4% п-изомера: вымораживание 8 часов смесью льда и соли (2:1). Хороший способ разделения, это восстановление п-изомера щелочным востановителем. П-толуидин может быть отделен благодаря своим основным свойствам. Лучше всего разделение достигается фракционной перегонкой с последующей кристаллизацией 11). Из фильтрата перегонкой в вакууме на 30 см колонке Вигре с электрическим обогревом выделяют о-изомер. Выход о-изомера 40 %. Температуры кипения о- и п-нитротолуола равны, соответственно, 96°С/9 мм. и 105°С /10 мм, температура плавления п-толуидина 52-54°С.
Физические свойства
Бензол и его ближайшие гомологи – бесцветные жидкости со специфическим запахом. Ароматические углеводороды легче воды и в ней не растворяются, однако легко растворяются в органических растворителях – спирте, эфире, ацетоне.
Бензол и его гомологи сами являются хорошими растворителями для многих органических веществ. Все арены горят коптящим пламенем ввиду высокого содержания углерода вих молекулах.
Физические свойства некоторых аренов представлены в таблице.
Таблица. Физические свойства некоторых аренов
|
Название |
Формула |
t°.пл., |
t°.кип., |
|
Бензол |
C 6 H 6 |
5,5 |
80,1 |
|
Толуол (метилбензол) |
С 6 Н 5 СH 3 |
95,0 |
110,6 |
|
Этилбензол |
С 6 Н 5 С 2 H 5 |
95,0 |
136,2 |
|
Ксилол (диметилбензол) |
С 6 Н 4 (СH 3) 2 |
||
|
орто- |
25,18 |
144,41 |
|
|
мета- |
47,87 |
139,10 |
|
|
пара- |
13,26 |
138,35 |
|
|
Пропилбензол |
С 6 Н 5 (CH 2) 2 CH 3 |
99,0 |
159,20 |
|
Кумол (изопропилбензол) |
C 6 H 5 CH(CH 3) 2 |
96,0 |
152,39 |
|
Стирол (винилбензол) |
С 6 Н 5 CH=СН 2 |
30,6 |
145,2 |
Бензол – легкокипящая ( t кип = 80,1°С), бесцветная жидкость, не растворяется в воде
Внимание! Бензол – яд, действует на почки, изменяет формулу крови (при длительном воздействии), может нарушать структуру хромосом.
Большинство ароматических углеводородов опасны для жизни, токсичны.
Получение аренов (бензола и его гомологов)
В лаборатории
1. Сплавление солей бензойной кислоты с твёрдыми щелочами
C 6 H 5 -COONa + NaOH t → C 6 H 6 + Na 2 CO 3
бензоат натрия
2. Реакция Вюрца-Фиттинга : (здесь Г – галоген)
С 6 H 5 -Г + 2 Na + R -Г → C 6 H 5 - R + 2 Na Г
С 6 H 5 -Cl + 2Na + CH 3 -Cl → C 6 H 5 -CH 3 + 2NaCl
В промышленности
- выделяют из нефти и угля методом фракционной перегонки, риформингом;
- из каменноугольной смолы и коксового газа
1. Дегидроциклизацией алканов с числом атомов углерода больше 6:
C 6 H 14 t , kat →C 6 H 6 + 4H 2
2. Тримеризация ацетилена (только для бензола) – р. Зелинского :
3С 2 H 2 600° C , акт. уголь →C 6 H 6
3. Дегидрированием циклогексана и его гомологов:
Советский академик Николай Дмитриевич Зелинский установил, что бензол образуется из циклогексана (дегидрирование циклоалканов
C 6 H 12 t, kat →C 6 H 6 + 3H 2

C 6 H 11 -CH 3 t , kat →C 6 H 5 -CH 3 + 3H 2
метилциклогексантолуол
4. Алкилирование бензола (получение гомологов бензола) – р Фриделя-Крафтса .
C 6 H 6 + C 2 H 5 -Cl t, AlCl3 →C 6 H 5 -C 2 H 5 + HCl
хлорэтан этилбензол

Химические свойства аренов
I . РЕАКЦИИ ОКИСЛЕНИЯ
1. Горение (коптящее пламя):
2C 6 H 6 + 15O 2 t →12CO 2 + 6H 2 O + Q
2. Бензол при обычных условиях не обесцвечивает бромную воду и водный раствор марганцовки
3. Гомологи бензола окисляются перманганатом калия (обесцвечивают марганцовку):
А) в кислой среде до бензойной кислоты
При действии на гомологи бензола перманганата калия и других сильных окислителей боковые цепи окисляются. Какой бы сложной ни была цепь заместителя, она разрушается, за исключением a -атома углерода, который окисляется в карбоксильную группу.
Гомологи бензола с одной боковой цепью дают бензойную кислоту:
Гомологи, содержащие две боковые цепи, дают двухосновные кислоты:
5C 6 H 5 -C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 6K 2 SO 4 + 12MnSO 4 +28H 2 O
5C 6 H 5 -CH 3 + 6KMnO 4 + 9H 2 SO 4 → 5C 6 H 5 COOH + 3K 2 SO 4 + 6MnSO 4 +14H 2 O
Упрощённо:
C 6 H 5 -CH 3 + 3O KMnO4 →C 6 H 5 COOH + H 2 O
Б) в нейтральной и слабощелочной до солей бензойной кислоты
C 6 H 5 -CH 3 + 2KMnO 4 → C 6 H 5 COO К + K ОН + 2MnO 2 + H 2 O
II . РЕАКЦИИ ПРИСОЕДИНЕНИЯ (труднее, чем у алкенов)
1. Галогенирование
C 6 H 6 +3Cl 2 h ν → C 6 H 6 Cl 6 (гексахлорциклогексан - гексахлоран)
2. Гидрирование
C 6 H 6 + 3H 2 t , Pt или Ni →C 6 H 12 (циклогексан)
3. Полимеризация
III . РЕАКЦИИ ЗАМЕЩЕНИЯ – ионный механизм(легче, чем у алканов)
1. Галогенирование -
a ) бензола
C 6 H 6 + Cl 2 AlCl 3 → C 6 H 5 -Cl + HCl (хлорбензол)
C 6 H 6 + 6Cl 2 t ,AlCl3 →C 6 Cl 6 + 6HCl ( гексахлорбензол )
C 6 H 6 + Br 2 t,FeCl3 → C 6 H 5 -Br + HBr ( бромбензол )
б) гомологов бензола при облучении или нагревании
По химическим свойствам алкильные радикалы подобны алканам. Атомы водорода в них замещаются на галоген по свободно-радикальному механизму. Поэтому в отсутствие катализатора при нагревании или УФ-облучении идет радикальная реакция замещения в боковой цепи. Влияние бензольного кольца на алкильные заместители приводит к тому, что замещается всегда атом водорода у атома углерода, непосредственно связанного с бензольным кольцом (a -атома углерода).
1) C 6 H 5 -CH 3 + Cl 2 h ν → C 6 H 5 -CH 2 -Cl + HCl
в) гомологов бензола в присутствии катализатора
C 6 H 5 -CH 3 + Cl 2 AlCl 3 → (смесь орта, пара производных) +HCl
2. Нитрование (с азотной кислотой)
C 6 H 6 + HO-NO 2 t, H2SO4 →C 6 H 5 -NO 2 + H 2 O
нитробензол - запах миндаля !
C 6 H 5 -CH 3 + 3HO-NO 2 t, H2SO4 → С H 3 -C 6 H 2 (NO 2) 3 + 3H 2 O2,4,6-тринитротолуол (тол, тротил)
Применение бензола и его гомологов
Бензол C 6 H 6 – хороший растворитель. Бензол в качестве добавки улучшает качество моторного топлива. Служит сырьем для получения многих ароматических органических соединений – нитробензола C 6 H 5 NO 2 (растворитель, из него получают анилин), хлорбензола C 6 H 5 Cl, фенола C 6 H 5 OH, стирола и т.д.
Толуол C 6 H 5 –CH 3 – растворитель, используется при производстве красителей, лекарственных и взрывчатых веществ (тротил (тол), или 2,4,6-тринитротолуол ТНТ).
Ксилолы C 6 H 4 (CH 3) 2 . Технический ксилол – смесь трех изомеров (орто -, мета - и пара -ксилолов) – применяется в качестве растворителя и исходного продукта для синтеза многих органических соединений.
Изопропилбензол C 6 H 5 –CH(CH 3) 2 служит для получения фенола и ацетона.
Хлорпроизводные бензола используют для защиты растений. Так, продукт замещения в бензоле атомов Н атомами хлора – гексахлорбензол С 6 Сl 6 – фунгицид; его применяют для сухого протравливания семян пшеницы и ржи против твердой головни. Продукт присоединения хлора к бензолу – гексахлорциклогексан (гексахлоран) С 6 Н 6 Сl 6 – инсектицид; его используют для борьбы с вредными насекомыми. Упомянутые вещества относятся к пестицидам – химическим средствам борьбы с микроорганизмами, растениями и животными.
Стирол C 6 H 5 – CH = CH 2 очень легко полимеризуется, образуя полистирол, а сополимеризуясь с бутадиеном – бутадиенстирольные каучуки.
ВИДЕО-ОПЫТЫ
Автор Л.А.ЦветковНитрование бензола может быть проведено с небольшими количествами исходных веществ, без выделения чистого продукта. Для получения нитробензола по уравнению:
С 6 Н 6 + HNO 3 à C 6 H 5 NO 2 + Н 2 О
необходима концентрированная азотная кислота (уд. вес. 1,4). Реакционная смесь при этом не должна нагреваться выше 50-60"С. При применении разбавленной кислоты реакция нитрования не идет; при повышении температуры начинается заметное образование динитробензола.
Из уравнения следует, что для реакции необходимы эквимолекулярные количества исходных веществ. Однако в таком случае реакция не дойдет до конца, так как выделяющаяся вода будет разбавлять азотную кислоту, и она потеряет нитрующее свойство. Следовательно, чтобы довести реакцию до конца, надо взять больше азотной кислоты, чем следует по теории. Но, чтобы реакция при этом не стала слишком бурной, азотную кислоту нужно растворить в концентрированной серной кислоте, которая не лишает азотную кислоту нитрующего действия и связывает выделяющуюся при реакции воду.
Чтобы предупредить возможность повышения температуры при реакции, не смешивают сразу все вещества, а к смеси кислот постепенно добавляют бензол. В небольшую колбочку наливают 8 мл концентрированной серной кислоты и 5 мл концентрированной азотной кислоты. Охлаждают смесь в струе воды. Затем к охлажденной смеси прибавляют небольшими порциями 4 мл бензола, постоянно встряхивая колбочку, чтобы достичь большего смешения нерастворяющихся друг в друге жидкостей (смесь кислот составляет нижний слой, бензол - верхний слой). После приливания всего бензола для достижения полноты реакции колбу закрывают пробкой с вертикальной трубкой (пары бензола летучи) и нагревают на предварительно нагретой до 60°С водяной бане
Время от времени колбу встряхивают, чтобы жидкости лучше перемешивались.
Продолжительность нагревания может определяться не столько необходимостью достижения полноты реакции, сколько наличием времени на уроке. При работе в кружке нагревание следует продолжать минут 30-40. Па уроке же удается продемонстрировать образование нитробензола после нагревания в течение 10 мин и даже вовсе без дополнительного нагревания, если реакция хорошо шла при приливании бензола к смеси кислот.
Нитробензол располагается слоем поверх смеси кислот. Выливают содержимое колбы в стакан с большим количеством воды. При этом кислоты растворяются в воде, нитробензол же собирается на дне стакана в виде тяжелой желтоватой жидкости. Если позволяет время, сливают часть жидкости с нитробензола и отделяют его с помощью делительной воронки.
При получении значительных количеств нитробензола и необходимости его очистки, нитробензол промывают водой, разбавленным (5-процентным) раствором щелочи, затем снова водой, разделяя всякий раз жидкости с помощью делительной воронки. После этого обезвоживают нитробензол, нагревая его с гранулированным хлоридом кальция, пока жидкость не станет прозрачной. Нагревание при этом необходимо, чтобы понизить вязкость нитробензола и достичь таким образом более полного контакта его с хлоридом кальция. Наконец, нитробензол может быть перегнан из небольшой колбочки с воздушным холодильником при температуре 204-207°С. Для того чтобы избежать разложения остатков динитробензола, не рекомендуется проводить перегонку досуха.
 Иван петрович павлов, открытия
Иван петрович павлов, открытия Мария значение имени - характер и судьба
Мария значение имени - характер и судьба Терентьев Фёдор Михайлович (Terent'ev Fedor Mikhailovich)
Терентьев Фёдор Михайлович (Terent'ev Fedor Mikhailovich)