Нитрование толуола азотной кислотой. Пиротехническая химия: Химия и технология бризантных взрывчатых веществ - Орлова Е.Ю
Нитрование ароматических углеводородов
а) Нитрование бензола (тяга!). В маленькой колбе смешивают 4 мл концентрированной серной кислоты ( =1,84) с 3 мл концентрированной азотной кислоты ( =1,4). К полученной смеси приливают по каплям 3 мл бензола, сильно встряхивая содержимое колбы (температура не должна подниматься выше 40 С), охлаждая в случае необходимости водой. Закрыв колбу пробкой с воздушным холодильником, нагревают ее 15 мин на водяной бане до 60 0 С, часто встряхивая. Затем реакционную смесь охлаждают и выливают в стакан с 20 мл ледяной воды, при этом образуются два слоя. Водный слой сливают, а выпавшее на дне масло (нитробензол) еще дважды промывают водой. После отделения от воды сырой нитробензол переливают в сухую пробирку, добавляют 2-3 кусочка прокаленного СаС1 2 и нагревают на водяной бане, пока нитробензол не станет прозрачным. Перегоняют нитробензол из маленькой колбы Вюрца или пробирки с нисходящей трубкой при 207-210 0 С. (Перегонять нитробензол досуха нельзя! Возможен взрыв!).
Напишите уравнение нитрования бензола. Какова роль серной кислоты при нитровании ароматических соединений? Объясните механизм нитрования ароматических соединений.
в) Нитрование толуола. При нитровании толуола возможно образование смеси орто- и пара - нитротолуолов. Контроль за процессом и идентификацию продуктов реакции можно проводить методом тонкослойной хроматографии. Хроматографирование проводят на силуфолевой пластинке, применяя четыреххлористый углерод в качестве элюента. Готовят нитрующую смесь из 3 мл концентрированной азотной кислоты ( =1,1) и 1 мл концентрированной серной кислоты. В пробирку с 2 мл толуола по каплям при охлаждении и встряхивании реакционной смеси добавляют нитрующую смесь. Затем пробирку закрывают пробкой с вертикальной трубкой и нагревают на водяной бане, часто встряхивая. Через 10 мин капилляром отбирают пробу реакционной массы и на стартовую линию силуфолевой пластинки наносят пробу раствора и «свидетелей» орто - и пара- нитротолуолов (в толуоле). Пластинку опускают в камеру с четыреххлористым углеродом и отмечают появление нитротолуолов.
Следующую пробу раствора отбирают через 40 мин, а третью - через 1 ч. Отмечают изменение состава реакционной среды.
Как меняется соотношение изомеров нитротолуолов в процессе реакции? Напишите уравнение реакции нитрования толуола. Рассмотрите механизм реакции. Объясните влияние строения ароматического соединения на легкость нитрования.
1 .. 40 > .. >> Следующая
Неочищенный тротил можно применять только для изготовления взрывчатых смесей, предназначенных к быстрому употреблению, например для взрывных работ. Тротил же, идущий для снаряжения боеприпасов, которые подлежат длительному хранению, должен быть обязательно подвергнут очистке.
ТЕХНОЛОГИЯ ПОЛУЧЕНИЯ ТРОТИЛА
Закономерности процесса нитрования
Нитрование толуола до мононитротолуола. При нитровании толуола до мононитротолуола получаются три изомера: орто-, пара- и мета, с преимущественным преобладанием орто-то-мера. .мета-Изомер образуется в относительно небольшом количестве, но так как при его дальнейшем нитровании получаются несимметричные трйнитротолуолы, то образование его нежелательно.
Основные работы по исследованию закономерностей реакции нитрования толуола до мононитротолуола были направлены главным образом на выявление условий наименьшего выхода лета-изомера. Когда непрерывные процессы нитрования стали доминирующими и назрел вопрос о рациональных конструкциях нитраторов, на"чалось изучение кинетики нитрования, в основном в гетеро-
Рис. 17. Растворимость толуола в серной кислоте различной концентрации при 55 °С.
65 70 75 80 85 90 Концентрация HzS047%
95
генной среде, что соответствует промышленным условиям. При этом исследовали растворимость толуола в серно-азотных кислотных смесях, распределение компонентов между слоями, влияние перемешивания и соотношения объемов органического и кислотного слоев на скорость реакции. Растворимость толуола в серной кислоте растет с повышением концентрации кислоты; до концентрации кислоты 80% растворимость толуола очень низка (рис. 17).
Скорость нитрования толуола в гетерогенных условиях резко зависит от интенсивности перемешивания и модуля ванны (отношение объемов минерального и органического слоев) (рис. 18).
Коэффициент распределения азотной кислоты между толуоль-ным и серно-кислотным слоями равен 0,066. Это-указывает на то, что азотная кислота при гетерогенном нитровании толуола лишь в незначительной степени переходит в органический слой и поэтому доля протекающей там реакции практически равна нулю.
Низкая растворимость толуола в серной кислоте умеренных концентраций, отсутствие перехода азотной кислоты в органический слой, а также резкая зависимость скорости реакции нитрования толуола от интенсивности перемешивания и объемной доли минерального слоя позволяют предположить, что реакция нитрования толуола в гетерогенных условиях протекает на поверхности раздела слоев. Скорость реакции в этом случае зависит от концентрации реагирующих компонентов^ на этой поверхности, которая в свою очередь определяется скоростью диффузии реагирующих компонентов из глубины слоя к поверхности раздела и скоростью отхода от нее продуктов реакции. Все это, а также состояние реагирующих компонентов зависит от температуры (рис. 19, с), концентрации кислотной смеси (рис. 19, б) и интенсивности перемешивания (рис. 18), причем скорость нитрования толуола в гетерогенных условиях ниже, чем в гомогенных (рис. 19,6).
Образование 5-6% л-нитротолуола при нитровании толуола в дальнейшем приводит к образованию 5-6% несимметричных тринитротолуолов, загрязняющих тротил. Температура затвердева-
Рис. 18. Влияние интенсивности перемешивания (а) и модуля ианны (6") на степень нитрования толуола.
ния тротила, содержащего несимметричные изомеры, снижается по следующей зависимости:
Г3 = (80,80 - 0.465С)
где С - содержание л-нитротолуола в исходном мононитротолуоле, %.
На изомерный состав мононитротолуола значительное влияние оказывает температура нитрования толуола (см. стр. 85). Исследование скорости нитрования толуола при 0 и 30 °С и определение изомерного состава позволило рассчитать в уравнении Аррениуса (см. стр. 54) коэффициент В для вступления нитрогруппы в различные положения относительно СН3-группы: В0 = 2,90 Вя, Вп = 2,70 Вм\ энергия активации для соответствующих положений равна: Е„ - Е0 = 3,83 кДж/(моль-°С), Е„ - ?„ = 4,61 кДж/(моль-°С). Из этих данных может быть сформулировано следующее правило" понижение температуры нитрования способствует увеличению выхода п-нитротолуола и уменьшению выхода о- и м-нитротолуолов.
Значительное влияние на выход л-нитротолуола оказывает фактор нитрующей активности кислотной смеси. Повышение Ф с 68 до 82% при нитровании толуола серно-азотными кислотными смесями при 55 °С снижает выход лета-изомера в 2,4 раза. На рис. 20 показано влияние температуры и фактора нитрующей активности кислотной смеси на выход л-нитротолуола.
Применение для нитрования толуола разбавленной азотной кислоты (70%-ной) приводит к образованию продуктов окисления, главным образом бензойной кислоты. Применение еще более разбавленной кислоты (32%-ной) при повышенной температуре (105 °С) вызывает нитрование боковой цепи, образуется фенил-нитрометан, который получается также и при нитровании толуола двуокисью азота.
-l-I-а,; ,„i, I_I I-1_¦ \ ¦_I
25 зо 40 60 № 70 во 64 ее ее ю 72 74
Твмперашура,°С Ф
Рис. 19. Зависимость скорости нитрования толуола от температуры (а) и фактора нитрующей активности Ф (6*):
Физические свойства
Бензол и его ближайшие гомологи – бесцветные жидкости со специфическим запахом. Ароматические углеводороды легче воды и в ней не растворяются, однако легко растворяются в органических растворителях – спирте, эфире, ацетоне.
Бензол и его гомологи сами являются хорошими растворителями для многих органических веществ. Все арены горят коптящим пламенем ввиду высокого содержания углерода вих молекулах.
Физические свойства некоторых аренов представлены в таблице.
Таблица. Физические свойства некоторых аренов
|
Название |
Формула |
t°.пл., |
t°.кип., |
|
Бензол |
C 6 H 6 |
5,5 |
80,1 |
|
Толуол (метилбензол) |
С 6 Н 5 СH 3 |
95,0 |
110,6 |
|
Этилбензол |
С 6 Н 5 С 2 H 5 |
95,0 |
136,2 |
|
Ксилол (диметилбензол) |
С 6 Н 4 (СH 3) 2 |
||
|
орто- |
25,18 |
144,41 |
|
|
мета- |
47,87 |
139,10 |
|
|
пара- |
13,26 |
138,35 |
|
|
Пропилбензол |
С 6 Н 5 (CH 2) 2 CH 3 |
99,0 |
159,20 |
|
Кумол (изопропилбензол) |
C 6 H 5 CH(CH 3) 2 |
96,0 |
152,39 |
|
Стирол (винилбензол) |
С 6 Н 5 CH=СН 2 |
30,6 |
145,2 |
Бензол – легкокипящая ( t кип = 80,1°С), бесцветная жидкость, не растворяется в воде
Внимание! Бензол – яд, действует на почки, изменяет формулу крови (при длительном воздействии), может нарушать структуру хромосом.
Большинство ароматических углеводородов опасны для жизни, токсичны.
Получение аренов (бензола и его гомологов)
В лаборатории
1. Сплавление солей бензойной кислоты с твёрдыми щелочами
C 6 H 5 -COONa + NaOH t → C 6 H 6 + Na 2 CO 3
бензоат натрия
2. Реакция Вюрца-Фиттинга : (здесь Г – галоген)
С 6 H 5 -Г + 2 Na + R -Г → C 6 H 5 - R + 2 Na Г
С 6 H 5 -Cl + 2Na + CH 3 -Cl → C 6 H 5 -CH 3 + 2NaCl
В промышленности
- выделяют из нефти и угля методом фракционной перегонки, риформингом;
- из каменноугольной смолы и коксового газа
1. Дегидроциклизацией алканов с числом атомов углерода больше 6:
C 6 H 14 t , kat →C 6 H 6 + 4H 2
2. Тримеризация ацетилена (только для бензола) – р. Зелинского :
3С 2 H 2 600° C , акт. уголь →C 6 H 6
3. Дегидрированием циклогексана и его гомологов:
Советский академик Николай Дмитриевич Зелинский установил, что бензол образуется из циклогексана (дегидрирование циклоалканов
C 6 H 12 t, kat →C 6 H 6 + 3H 2

C 6 H 11 -CH 3 t , kat →C 6 H 5 -CH 3 + 3H 2
метилциклогексантолуол
4. Алкилирование бензола (получение гомологов бензола) – р Фриделя-Крафтса .
C 6 H 6 + C 2 H 5 -Cl t, AlCl3 →C 6 H 5 -C 2 H 5 + HCl
хлорэтан этилбензол

Химические свойства аренов
I . РЕАКЦИИ ОКИСЛЕНИЯ
1. Горение (коптящее пламя):
2C 6 H 6 + 15O 2 t →12CO 2 + 6H 2 O + Q
2. Бензол при обычных условиях не обесцвечивает бромную воду и водный раствор марганцовки
3. Гомологи бензола окисляются перманганатом калия (обесцвечивают марганцовку):
А) в кислой среде до бензойной кислоты
При действии на гомологи бензола перманганата калия и других сильных окислителей боковые цепи окисляются. Какой бы сложной ни была цепь заместителя, она разрушается, за исключением a -атома углерода, который окисляется в карбоксильную группу.
Гомологи бензола с одной боковой цепью дают бензойную кислоту:
Гомологи, содержащие две боковые цепи, дают двухосновные кислоты:
5C 6 H 5 -C 2 H 5 + 12KMnO 4 + 18H 2 SO 4 → 5C 6 H 5 COOH + 5CO 2 + 6K 2 SO 4 + 12MnSO 4 +28H 2 O
5C 6 H 5 -CH 3 + 6KMnO 4 + 9H 2 SO 4 → 5C 6 H 5 COOH + 3K 2 SO 4 + 6MnSO 4 +14H 2 O
Упрощённо:
C 6 H 5 -CH 3 + 3O KMnO4 →C 6 H 5 COOH + H 2 O
Б) в нейтральной и слабощелочной до солей бензойной кислоты
C 6 H 5 -CH 3 + 2KMnO 4 → C 6 H 5 COO К + K ОН + 2MnO 2 + H 2 O
II . РЕАКЦИИ ПРИСОЕДИНЕНИЯ (труднее, чем у алкенов)
1. Галогенирование
C 6 H 6 +3Cl 2 h ν → C 6 H 6 Cl 6 (гексахлорциклогексан - гексахлоран)
2. Гидрирование
C 6 H 6 + 3H 2 t , Pt или Ni →C 6 H 12 (циклогексан)
3. Полимеризация
III . РЕАКЦИИ ЗАМЕЩЕНИЯ – ионный механизм(легче, чем у алканов)
1. Галогенирование -
a ) бензола
C 6 H 6 + Cl 2 AlCl 3 → C 6 H 5 -Cl + HCl (хлорбензол)
C 6 H 6 + 6Cl 2 t ,AlCl3 →C 6 Cl 6 + 6HCl ( гексахлорбензол )
C 6 H 6 + Br 2 t,FeCl3 → C 6 H 5 -Br + HBr ( бромбензол )
б) гомологов бензола при облучении или нагревании
По химическим свойствам алкильные радикалы подобны алканам. Атомы водорода в них замещаются на галоген по свободно-радикальному механизму. Поэтому в отсутствие катализатора при нагревании или УФ-облучении идет радикальная реакция замещения в боковой цепи. Влияние бензольного кольца на алкильные заместители приводит к тому, что замещается всегда атом водорода у атома углерода, непосредственно связанного с бензольным кольцом (a -атома углерода).
1) C 6 H 5 -CH 3 + Cl 2 h ν → C 6 H 5 -CH 2 -Cl + HCl
в) гомологов бензола в присутствии катализатора
C 6 H 5 -CH 3 + Cl 2 AlCl 3 → (смесь орта, пара производных) +HCl
2. Нитрование (с азотной кислотой)
C 6 H 6 + HO-NO 2 t, H2SO4 →C 6 H 5 -NO 2 + H 2 O
нитробензол - запах миндаля !
C 6 H 5 -CH 3 + 3HO-NO 2 t, H2SO4 → С H 3 -C 6 H 2 (NO 2) 3 + 3H 2 O2,4,6-тринитротолуол (тол, тротил)
Применение бензола и его гомологов
Бензол C 6 H 6 – хороший растворитель. Бензол в качестве добавки улучшает качество моторного топлива. Служит сырьем для получения многих ароматических органических соединений – нитробензола C 6 H 5 NO 2 (растворитель, из него получают анилин), хлорбензола C 6 H 5 Cl, фенола C 6 H 5 OH, стирола и т.д.
Толуол C 6 H 5 –CH 3 – растворитель, используется при производстве красителей, лекарственных и взрывчатых веществ (тротил (тол), или 2,4,6-тринитротолуол ТНТ).
Ксилолы C 6 H 4 (CH 3) 2 . Технический ксилол – смесь трех изомеров (орто -, мета - и пара -ксилолов) – применяется в качестве растворителя и исходного продукта для синтеза многих органических соединений.
Изопропилбензол C 6 H 5 –CH(CH 3) 2 служит для получения фенола и ацетона.
Хлорпроизводные бензола используют для защиты растений. Так, продукт замещения в бензоле атомов Н атомами хлора – гексахлорбензол С 6 Сl 6 – фунгицид; его применяют для сухого протравливания семян пшеницы и ржи против твердой головни. Продукт присоединения хлора к бензолу – гексахлорциклогексан (гексахлоран) С 6 Н 6 Сl 6 – инсектицид; его используют для борьбы с вредными насекомыми. Упомянутые вещества относятся к пестицидам – химическим средствам борьбы с микроорганизмами, растениями и животными.
Стирол C 6 H 5 – CH = CH 2 очень легко полимеризуется, образуя полистирол, а сополимеризуясь с бутадиеном – бутадиенстирольные каучуки.
ВИДЕО-ОПЫТЫ
Нитрование
Реакция нитрования является одной из наиболее изученных реакций ароматического замещения. Для препаративных целей нитрование, как правило, проводят смесью концентрированных азотной и серной кислот, так называемой нитрующей смесью . На первой стадии реакции происходит образование иона нитрония + NO 2 , который и является электрофильным агентом:
HO-NO 2 + H 2 SO 4 H 2 O + -NO 2 + HSO 4 -
H 2 O + -NO 2 + H 2 SO 4 H 3 O + + HSO 4 - + + NO 2
Наличие иона нитрония в этом растворе подтверждено спектроскопически. Азотная кислота в концентрированной серной кислоте практически нацело превращается в нитроний-катион. Незначительная эффективность самой азотной кислоты в реакции нитрования бензола объясняется низким содержанием иона + NO 2 .
B качестве нитрующих агентов используются также другие системы, в которых генерируется либо катион + NO 2 , либо соединение общей формулы NO2-Y где Y - хорошая уходящая группа. Некоторые из таких систем, нашедшие наибольшее применение, представлены в таблице 1 в порядке увеличения их активности.
Таблица 1. Нитрующие реагенты.
| Нитрующий реагент | Метод генерации | Арены, подвергающиеся нитрованию |
| Азотная кислота HO-NO2 |
Фенолы, эфиры фенолов, бифенил | |
| Ацетилнитрат CH 3 C(O)-O-NO 2 | СН 3 СООН + HNO 3 (СН 3 СО) 2 О + HNO 3 | Бензол, алкилбензолы |
| Диоксид азота N 2 O 4 (O=N-O-NO 2) |
Бензол, алкилбензолы | |
| Нитрующая смесь | H 2 SO 4 конц + HNO 3 | Бензол, алкилбензолы, галогенбензолы, бензойная кислота, нитробензол, нафталин |
| Хлорид нитрония Сl-NO 2 |
Бензол, алкилбензолы, нитробензол, | |
| Тетрафторборат нитрония BF 4 -+ NO 2 | HF . 2BF 3 + HNO 3 | Динитробензол |
На примере реакции нитрования алкилбензолов отчетливо прослеживается влияние пространственных факторов на направление электрофильного замещения. Так, при нитровании толуола (метилбензола) орто -изомер образуется в качестве основного продукта, а при переходе к этил-, изо -пропил- и особенно к трет -бутил-бензолу его выход существенно уменьшается (см. табл. 2).
Таблица 2. Влияние пространственых факторов на соотношение орто-, пара-изомеров в реакции нитрования (NO 2 +)
Замещение в положения, % |
|||
C 6 H 5 -C 2 H 5 |
|||
C 6 H 5 -CH(CH 3) 2 |
|||
C 6 H 5 -C(CH 3) 3 |
|||
При изучении нитрования алкилбензолов было обнаружено так называемое ипсо-замещение , когда электрофильная атака протекает по тому атому углерода бензольного кольца, которое уже содержит заместитель, например:

В отличие от нитрования, при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены, например, Cl 2 и Br 2 ,(прим.35) могут легко атаковать активированное ароматическое ядро (например, фенола), но не способны реагировать с бензолами и алкилбензолами (фотохимическая активация может, однако, в последнем случае привести к протеканию радикального замещения в боковую цепь; см. раздел IV.3). Для поляризации атакующей молекулы галогена необходим катализ кислотами Льюиса , такими как AlCl 3 , FeBr 3 , и т.п.; при этом в молекуле галогена появляется так называемый "электрофильный конец" (энергия же, требующаяся для образования катиона Наl + существенно выше). Тем самым электрофильное замещение существенно облегчается:

Галогенирование протекает очень энергично, если использовать реагенты, в которых галоген в результате поляризации имеет сильный положительный заряд или даже существует как катион. Так, очень инертный мета -динитробензол можно пробромировать бромом в концентрированной серной кислоте в присутствии сульфата серебра. Предполагают, что в этом случае промежуточно образуется бром-катион:
2Br 2 + Ag 2 SO 4 2Br + + 2AgBr + SO 4 2-
Реакционная способность элементарного иода в реакциях электрофильного замещения в ароматическом ядре незначительна, так что прямое иодирование возможно только в случае фенола и ароматических аминов. Иодирование других ароматических соединений проводят в присутствии окислителя (обычно, азотной кислоты). Считается, что в этих условиях роль электрофильного агента играет ион I- + OH 2 .

Для галогенирования аренов можно применять также смешанные галогены , например, монохлорид брома (BrCl) или иода (ICl):

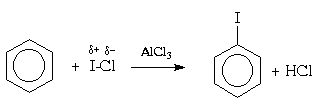
Галогенирование in vivo . В качестве примера электрофильного ароматического галогенирования, протекающего в живых организмах, можно привести реакцию иодирования -аминокислоты - тирозина в ходе биосинтеза иодсодержащих гормонов щитовидной железы до 3-иодтирозина и далее до 3,5-дииодтирозина:

Детали механизма сульфирования исследованы менее подробно по сравнению с нитрованием и галогенированием. Сам бензол сульфируется довольно медленно горячей концентрированной серной кислотой, но быстро - олеумом, SO 3 в инертных растворителях или комплексом SO 3 с пиридином. Природа электрофильной частицы зависит от условий реакции, но, вероятно, это всегда SO 3 , или в свободном состоянии, или связанный с "носителем", например, в виде H 2 SO 4 . SO 3 (H 2 S 2 O 7) в серной кислоте. Небольшие количества SO 3 образуются в H 2 SO 4:
2H 2 SO 4 SO 3 + H 3 O + + HSO 4 -
Атаку ароматического субстрата осуществляет атом серы поскольку он сильно положительно поляризован, то есть электронодефицитен:

Сульфирование является обратимым процессом. Это имеет практическое значение: при обработке сульфокислот водяным паромпроисходит замещение группы SO 3 Н на водород. Таким образом, можно ввести группу SO 3 Н как заместитель, ориентирующий требуемым образом последующие реакции (см. раздел IV.1.Б), а затем ее отщепить. Некоторые интересные особенности имеет сульфирование нафталина (см. раздел IV.1.Г).
Подобно галогенам, алкилгалогениды могут быть так сильно поляризованы кислотами Льюиса (хлоридами алюминия и цинка, трифторидом бора и др.), что они становятся способными к электрофильному замещению в ароматическом ядре:
R-Cl + AlCl 3 R +... Cl ...- AlCl 3 R + AlCl 4 -
Кроме алкилгалогенидов, источниками карбокатионов для галогенирования ароматических соединений могут быть алкены или спирты. При этом необходимо присутствие протонной кислоты, чтобы протонировать алкен или спирт. В случае спиртов требуется добавка не менее чем эквимольного количества кислоты (так как вода, выделяющаяся в ходе реакции, дезактивирует эквимольное количество катализатора), тогда как в реакциях с участием алкилгалогенидов и алкенов достаточно добавлять незначительное количество катализатора.
В лаборатории алкилирование по Фриделю-Крафтсу имеет ограниченное применение, так как обычно при этой реакции образуются смеси продуктов, что обусловлено рядом причин:
1) Образующийся продукт алкилирования легче вступает в реакции электрофильного ароматического замещения, чем исходное соединение (Alk - электронодонорная группа), поэтому дальше преимущественно алкилируется продукт. Если хотят получить продукты моноалкилирования, то необходимо брать большой избыток ароматического соединения.
2) Как и сульфирование, реакция алкилирования по Фриделю-Крафтсу обратима (см. также раздел IV.1.Г).
3) Даже в мягких условиях первичные и вторичные алкилгалогениды дают преимущественно вторичные или третичные алкиларены соответственно, поскольку алкилирование происходит в условиях, приближающихся к S N 1 реакции.(прим.37) Перегруппировки можно избежать, если работать при низких температурах.
Ацилирование ароматических соединений по Фриделю-Крафтсу является важнейшим методом синтеза жирноароматических кетонов. Производные карбоновых кислот, такие как ацилгалогениды и ангидриды, имеют полярную карбонильную группу и в принципе способны к электрофильному замещению в ароматических системах:

Электрофильная активность этих соединений, однако, невелика, и должна быть повышена действием кислот Льюиса. При этом кислотный катализатор, как правило, атакует атом кислорода карбонильного соединения и, смещая электронную плотность, повышает положительный заряд соседнего атома углерода. В результате образуется поляризованный комплекс (а в пределе - ацилкатион), действующий как электрофил:

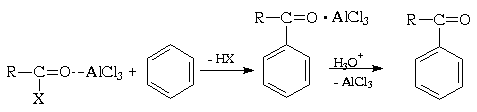
Важное отличие реакции ацилирования ацилгалогенидами от реакции алкилирования алкилгалогенидами состоит в том, что в первой из этих реакций требуется более 1 моль кислоты Льюиса , тогда как во второй необходимо только каталитические количество. Это обусловлено тем, что кислота Льюиса образует комплекс как с ацилирующим производным карбоновой кислоты, так и с кетоном - продуктом реакции. При взаимодействии с ангидридами получающаяся кислота связывает еще моль моль катализатора, так что в целом его необходимо по крайней мере два моль. В каждом случае по окончании реакции образовавшийся комплекс кетона с хлоридом алюминия (или другой кислотой Льюиса) должен быть гидролитически разрушен (соляной кислотой со льдом).
Полиацилирования не наблюдается, поскольку образующийся кетон значительно менее реакционноспособен, чем исходное соединение (см. раздел IV.1.Б). Поэтому алкилбензолы часто предпочитают получать не прямым алкилированием, а ацилированием по Фриделю-Крафтсу с последующим восстановлением. Ароматические соединения с сильнодезактивирующими заместителями, например, нитро- или циано- группами, также не ацилируются по Фриделю-Крафтсу.
Контрольные задачи
2. Изобразите диаграмму потенциальной энергии
для реакции электрофильного ароматического
замещения, в которой медленной стадией является
образование
-комплекса (например,
нитрование бензола борфторидом нитрония;
см. раздел IV.1.A).
3. Какой продукт преимущственно образуется при бромировании: а)пара -нитротолуола; б) мета -нитробензолсульфокислоты; в) орто -нитрофенола.
4. Адреналин (1-(3",4"-дигидроксифенил)-2-метиламиноэтанол)- первый гормон, выделенный из мозгового вещества надпочечников, в настоящее время синтезируюют в три стадии из пирокатехина. Напишите уравнение первой стадии этого синтеза - реакции ацилирования пирокатехина (1,2-дигидроксибензола) хлорангидридом хлоруксусной киcлоты и объясните механизм).
5. Одной их качественных реакций на белки является ксантопротеиновая реакция, указывающая на присутствие ароматических -аминокислот. Она заключается в обработке белка азотной кислотой при нагревании. Напишите уравнение ксантопротеиновой реакции с тирозином (см. раздел I), образовавшимся в результате гидролиза белка.
Получение через диазосоединение
Методы этой группы гораздо менее многочисленны, но отличаются высокими выходами, низкими содержаниями примесей побочных продуктов, простотой и разнообразием в исполнении.
Наиболее простым и надежным способом этой группы является проведение реакции Зандмейера. Можно привести 2 примера различного проведения только этой стадии:

2.3 Другие методы

PhBr + TfOMe, антраниловая кислота в реакции Бородина- Хунсдикера, реакция о-дибромбензола и MeMgBr и тд. - имеют преимуществ по сравнению с другими методами и отличаются более низкой препаративной ценностью, хотя и представляют интерес.
Выбранный путь синтеза - анализ, описание методик выполнения стадий эксперимента
Основным критерием выбора того или иного метода, подробно описанного выше, является надежность и доступность. Этому отвечает путь толуол - нитротолуол - о-толуидин - о-бромтолуол.
Нитрование толуола
В трехгорлую колбу на 250 мл, снабженную мешалкой, капельной воронкой, внутренним термометром (прибор не должен быть герметичен) помещают 0,15 моль ароматического нитросоединения. Затем медленно, при хорошем перемешивании и охлаждении баней со льдом добавляют нитрующую смесь, предварительно охлажденную до по меньшей мере до 10 °C, температура реакционной смеси должна находится в интервале 5-10 °C.
Далее при комнотной температуре перемешивают ещё 2-3 часа. После этого реакционную смесь осторожно выливают в 300 мл ледяной воды и хорошо перемешивают. Отделяют органический слой, водный экстрагируют эфиром. Объединённые органические вытяжки промывают водой, 2 н. раствором бикарбоната натрия до нейтральной реакции, потом опять водой. обогревателем. Вытяжки высушивают над CaCl 2 и перегоняют. П-изомер вымораживают смесью льда и соли, промывают небольшим количеством холодного петролейного эфира. (Тщательное разделение довольно, этот метод оставляет около 4% п-изомера: вымораживание 8 часов смесью льда и соли (2:1). Хороший способ разделения, это восстановление п-изомера щелочным востановителем. П-толуидин может быть отделен благодаря своим основным свойствам. Лучше всего разделение достигается фракционной перегонкой с последующей кристаллизацией 11). Из фильтрата перегонкой в вакууме на 30 см колонке Вигре с электрическим обогревом выделяют о-изомер. Выход о-изомера 40 %. Температуры кипения о- и п-нитротолуола равны, соответственно, 96°С/9 мм. и 105°С /10 мм, температура плавления п-толуидина 52-54°С.
 Education in Great Britain - Образование в Великобритании (5), устная тема по английскому языку с переводом
Education in Great Britain - Образование в Великобритании (5), устная тема по английскому языку с переводом Иван петрович павлов, открытия
Иван петрович павлов, открытия Мария значение имени - характер и судьба
Мария значение имени - характер и судьба